
Contents
- 1 動脈圧(arterial pressure)の長期調節機構
- 2 動脈圧の制御における腎・体液系(RENAL–BODY FLUID SYSTEM FOR ARTERIAL PRESSURE CONTROL)
- 3 圧利尿(pressure diuresis)の定量化と動脈圧(arterial pressure)制御の基礎
- 4 動脈圧制御における腎・体液系(Renal–Body Fluid System)の実験
- 5 腎・体液機構(Renal–Body Fluid Mechanism)は長期的な動脈圧制御にほぼ無限のフィードバックゲインを提供する
- 6 長期的な動脈圧(arterial pressure)の2つの主要な決定因子
- 7 慢性的な腎排出曲線は急性曲線よりもはるかに急峻
- 8 体液摂取(fluid intake)と腎機能(renal function)が変化しない場合、全末梢抵抗(total peripheral resistance)の増加が動脈圧(arterial pressure)の長期レベルを上昇させない
- 9 体液量(fluid volume)の増加は心拍出量(cardiac output)または全末梢抵抗(total peripheral resistance)を増加させることで動脈圧(arterial pressure)を上昇させる
- 10 動脈圧(arterial pressure)調節における腎・体液スキーマにおける塩(NaCl)の重要性
- 11 腎機能障害による慢性高血圧(CHRONIC HYPERTENSION)
- 12 腎質量減少と塩摂取増加による実験的容量負荷性高血圧(volume-loading hypertension)
- 13 容量負荷性高血圧の発達中の循環機能の連続的変化
- 14 腎臓がなく人工腎(artificial kidney)で維持されている患者における容量負荷性高血圧(volume-loading hypertension)
- 15 過剰なアルドステロン(aldosterone)による高血圧
- 16 動脈圧(arterial pressure)制御におけるレニン–アンジオテンシン系(renin-angiotensin system)の役割 ※これはマスタしなといけない!
- 17 レニン–アンジオテンシン系の構成要素
- 18 レニン–アンジオテンシン系の圧受容器圧反応の迅速さと強さ
- 19 アンジオテンシンIIは塩と水の腎保持を引き起こす—動脈圧(arterial pressure)の長期制御のための重要な手段
- 20 アンジオテンシンIIの塩と水の腎保持を引き起こす直接的な腎作用のメカニズム
- 21 アンジオテンシンIIはアルドステロン分泌を刺激して腎臓での塩と水の保持を増加させる
- 22 アンジオテンシンIIによる動脈圧(arterial pressure)の変化の定量的分析
- 23 レニン–アンジオテンシン系が塩摂取量の大きな変動にもかかわらず正常な動脈圧を維持する役割
- 24 レニン分泌腫瘍または腎虚血(renal ischemia)による高血圧
- 25 一腎ゴールドブラット高血圧(One-Kidney Goldblatt Hypertension)
- 26 二腎ゴールドブラット高血圧(Two-Kidney Goldblatt Hypertension)
- 27 レニンを慢性的に分泌する病変腎による高血圧
- 28 容量負荷と血管収縮の組み合わせによるその他のタイプの高血圧
- 29 大動脈縮窄(Coarctation of the Aorta)による上半身の高血圧
- 30 大動脈縮窄による高血圧における自己調節の役割
- 31 妊娠中毒症(Preeclampsia, Toxemia of Pregnancy)による高血圧
- 32 神経性高血圧(Neurogenic Hypertension)
- 33 高血圧の遺伝的原因(Genetic Causes of Hypertension)珍しい
- 34 一次性(本態性)高血圧(PRIMARY (ESSENTIAL) HYPERTENSION)(※原因不明という意味)
- 35 本態性高血圧における動脈圧制御のグラフィック分析(Graphic Analysis of Arterial Pressure Control in Essential Hypertension)
- 36 本態性高血圧の治療(Treatment of Essential Hypertension)
- 37 動脈圧制御のための統合された多面的システムの概要
- 38 数秒または数分以内に作用する動脈圧制御メカニズム
- 39 数分後に作用する動脈圧制御メカニズム
- 40 動脈圧の長期的な調整メカニズム
動脈圧(arterial pressure)の長期調節機構
第18章で説明した動脈圧の迅速な調節機構に加えて、体には週単位や月単位で動脈圧を調節する強力な機構も存在します。この長期的な動脈圧の調節は、体液量(body fluid volume)の恒常性(homeostasis)と密接に関連しており、それは体液の摂取と排出のバランスによって決まります。長期的な生存のためには、体液の摂取と排出が正確にバランスされる必要があり、その役割を果たしているのが、複数の神経系(nervous)とホルモン制御(hormonal controls)、および腎臓(kidneys)の塩と水の排泄を調節する局所制御システム(local control systems)です。本章では、長期的な血圧(blood pressure)調節において主要な役割を果たすこれらの腎・体液系(renal–body fluid systems)について説明します。
動脈圧の制御における腎・体液系(RENAL–BODY FLUID SYSTEM FOR ARTERIAL PRESSURE CONTROL)
動脈圧の制御における腎・体液系はゆっくりとではあるが強力に作用します。血液量(blood volume)が増加し、血管の容量(vascular capacitance)が変化しない場合、動脈圧も上昇します。この上昇した圧力により、腎臓が過剰な量を排泄し、圧力を正常に戻すという仕組みです。
動物の発生における系統発生(phylogenetic history)の中で、この圧力制御のための腎・体液系は原始的な機構です。この機構は脊椎動物(vertebrates)の中でも最も下等な部類の一つであるヌタウナギ(hagfish)において完全に作動しています。この動物の動脈圧はわずか8~14 mmHgで、その圧力は血液量にほぼ直接比例して増加します。ヌタウナギは常に海水を飲み、それが血液に吸収されて血液量と血圧が増加します。しかし、圧力が高くなりすぎると、腎臓が過剰な量を尿として排泄し、圧力を軽減します。圧力が低い場合、腎臓は摂取されたより少ない量の体液を排泄します。したがって、ヌタウナギが水を飲み続けるため、細胞外液量(extracellular fluid volume)、血液量、および圧力は再び高いレベルに達します。
この原始的な圧力制御機構は時代を経て生き残ってきましたが、塩と水の排泄を調節する神経系、ホルモン、および局所制御システムが追加されました。人間においても、腎臓の水と塩の排出は、ヌタウナギと同じくらい、またはそれ以上に圧力変化に敏感です。実際、人間の動脈圧がわずか数mmHg上昇するだけで、腎臓の水の排出量が倍増し、この現象を圧利尿(pressure diuresis)と呼び、同様に塩の排出量も倍増し、これを圧ナトリウム利尿(pressure natriuresis)と呼びます。
ヌタウナギと同様に、人間においても、動脈圧の制御における腎・体液系は長期的な動脈圧制御の基本的な機構です。しかし進化の過程で、このシステムにはさまざまな改良が加えられ、その制御がより正確になりました。特に重要な改良の一つが、後述するレニン–アンジオテンシン機構(renin-angiotensin mechanism)の追加です。
圧利尿(pressure diuresis)の定量化と動脈圧(arterial pressure)制御の基礎
図19-1は、分離された腎臓(isolated kidney)による塩と水の排泄に対する異なる動脈圧レベルの概ね平均的な影響を示しており、圧力が上昇するにつれて尿排出が著しく増加することを示しています。この増加した尿排出は圧利尿(pressure diuresis)という現象です。この図の曲線は腎尿排出曲線(renal urinary output curve)または腎機能曲線(renal function curve)と呼ばれます。人間では、動脈圧が50 mmHgのとき、尿排出はほとんどゼロです。100 mmHgで正常値となり、200 mmHgで正常の4〜6倍になります。さらに、動脈圧の上昇は尿量の増加だけでなく、ナトリウム(sodium)の排出量もほぼ同程度に増加させ、これを圧ナトリウム利尿(pressure natriuresis)と呼びます。
動脈圧制御における腎・体液系(Renal–Body Fluid System)の実験
図19-2は、イヌを用いた実験の結果を示しており、この実験では血圧(blood pressure)制御のためのすべての神経反射機構(nervous reflex mechanisms)が最初にブロックされました。その後、約400 mlの血液を静脈内に注入して動脈圧を急激に上昇させました。心拍出量(cardiac output)が正常の約2倍に急増し、平均動脈圧(mean arterial pressure)が静止レベルから115 mmHg上昇して205 mmHgになったことがわかります。この上昇した動脈圧が尿排出に及ぼす影響は中間の曲線で示されており、尿排出が12倍に増加しました。この非常に多量の体液が尿として失われることで、心拍出量と動脈圧の両方がその後の1時間で正常に戻りました。このように、腎臓は高い動脈圧に応じて体から過剰な体液を排泄し、動脈圧を正常に戻す極端な能力を持っていることがわかります。
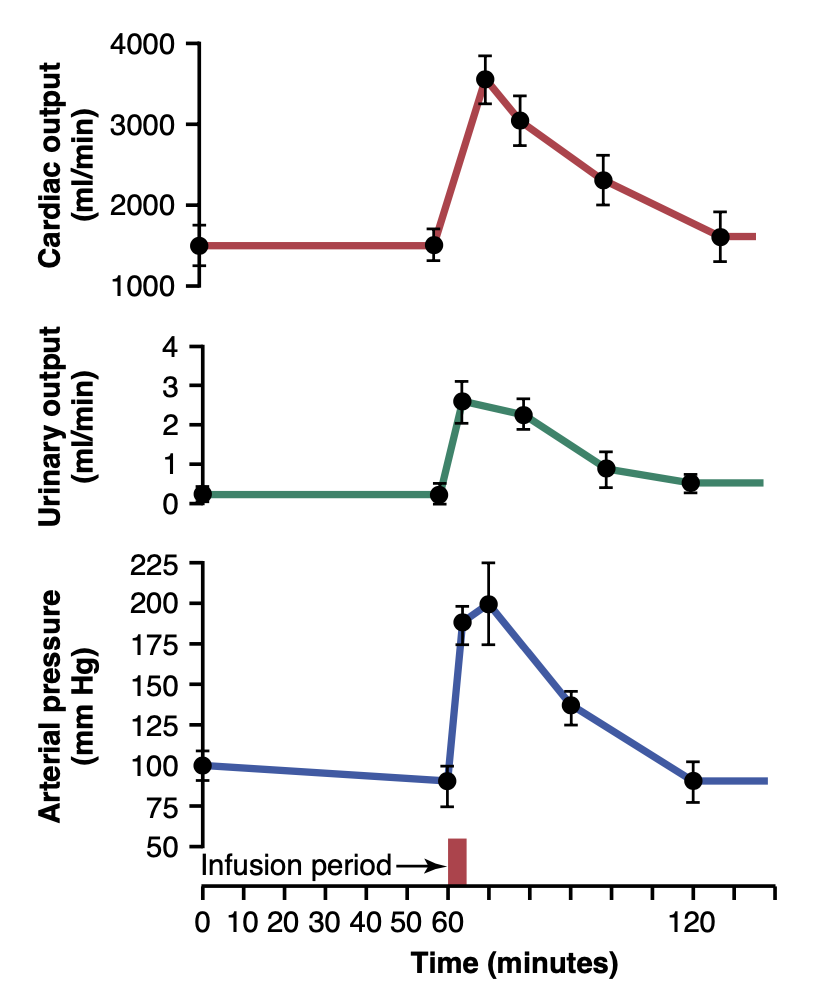
Figure 19-2. Increases in cardiac output, urinary output, and arterial pressure caused by increased blood volume in dogs whose nervous pressure control mechanisms had been blocked. This figure shows return of arterial pressure to normal after about 1 hour of fluid loss into the urine. (Courtesy Dr. William Dobbs.)
腎・体液機構(Renal–Body Fluid Mechanism)は長期的な動脈圧制御にほぼ無限のフィードバックゲインを提供する
図19-1は次の関係を示しています。(1) 上昇する動脈圧に応じた水と塩の腎排出曲線、および (2) 水と塩の純摂取量(net intake)を表す線です。長期間にわたり、水と塩の排出量は摂取量と等しくなければなりません。さらに、図19-1のグラフで排出量と摂取量が等しくなる唯一の点が、2つの曲線が交差する平衡点(equilibrium point、点A)です。動脈圧が平衡点を上回るか下回る場合に何が起こるか見てみましょう。
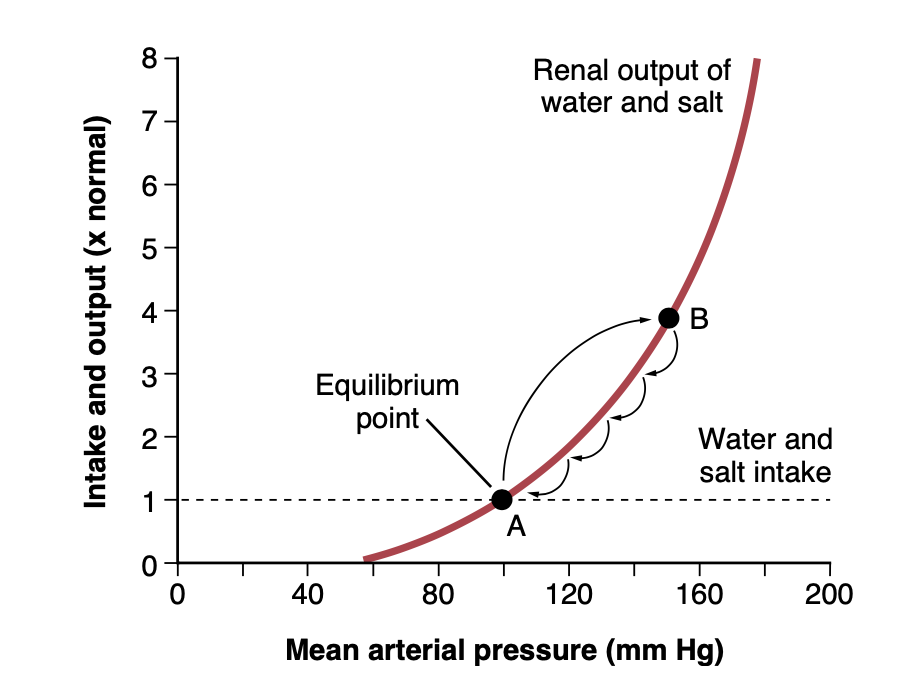
Figure 19-1. A typical arterial pressure–renal urinary output curve measured in a perfused isolated kidney, showing pressure diuresis when the arterial pressure rises above normal (point A) to approximately 150 mm Hg (point B). The equilibrium point A describes the level to which the arterial pressure will be regulated if intake is not altered. (Note that the small portion of the salt and water intake that is lost from the body through nonrenal routes is ignored in this and similar figures in this chapter.)
まず、動脈圧が150 mmHg(点B)に上昇したと仮定します。このレベルでは、水と塩の腎排出量は摂取量のほぼ3倍です。したがって、体は体液を失い、血液量が減少し、動脈圧が低下します。さらに、この体液の負のバランスは、圧力が平衡レベルまで完全に低下するまで止まりません。動脈圧が平衡レベルよりわずかに高い場合でも、水と塩の排出が摂取をわずかに上回り、圧力はわずかに低下し続け、最終的には平衡点に戻ります。
動脈圧が平衡点を下回る場合、水と塩の摂取量は排出量を上回ります。したがって、体液量が増加し、血液量が増加し、動脈圧が再び平衡点に戻るまで上昇します。この動脈圧が常に平衡点に戻る現象は、腎・体液機構による動脈圧制御のほぼ無限のフィードバックゲイン原理(near-infinite feedback gain principle)として知られています。
長期的な動脈圧(arterial pressure)の2つの主要な決定因子
図19-1では、長期的な動脈圧のレベルを決定する2つの基本的な長期要因も確認できます。図19-1に示されている塩と水の腎排出(renal output)と塩と水の摂取量(intake)を表す2つの曲線がそのままの位置にある限り、平均動脈圧レベルは最終的に100 mmHgに調整されます。これは図の平衡点(equilibrium point)で示されている圧力レベルです。さらに、この平衡点の圧力を100 mmHgから変える方法は2つだけです。一つは塩と水の腎排出曲線の圧力レベルを変えること、もう一つは塩と水の摂取量のレベルを変えることです。したがって、長期的な動脈圧レベルの2つの主要な決定因子を簡単に表現すると、次のようになります。
- 水と塩の腎排出曲線の圧力シフトの程度()
- 水と塩の摂取量のレベル()
動脈圧制御におけるこれら2つの決定因子の作用は図19-3に示されています。図19-3Aでは、腎臓の異常により腎排出曲線が高圧方向に50 mmHgシフトしています(右に移動)。平衡点も通常より50 mmHg高い位置にシフトしていることに注目してください。したがって、腎排出曲線が新しい圧力レベルにシフトすれば、動脈圧も数日以内にその新しい圧力レベルに従うことがわかります。
図19-3Bは、塩と水の摂取レベルの変化が動脈圧にどのように影響するかを示しています。この場合、摂取レベルが4倍に増加し、平衡点が通常レベルより60 mmHg高い160 mmHgにシフトしました。逆に、摂取レベルの減少は動脈圧を低下させます。
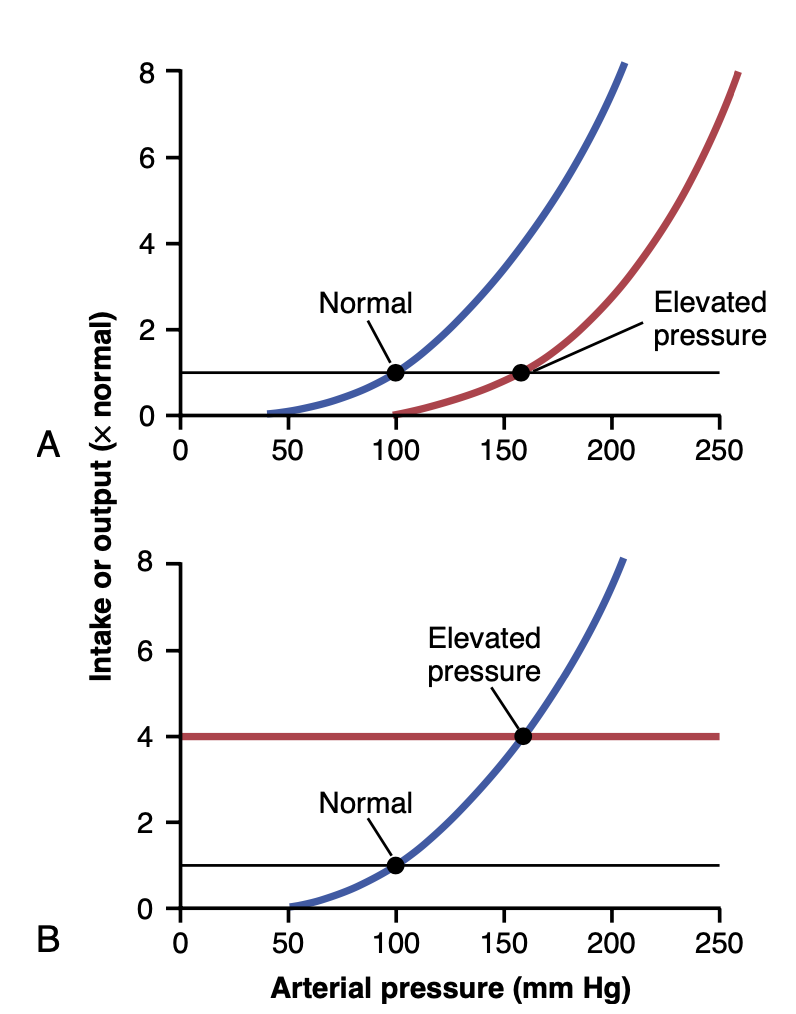
Figure 19-3. Two ways in which the arterial pressure can be in- creased. A, By shifting the renal output curve in the right-hand direc- tion toward a higher pressure level or by increasing the intake level of salt and water (B).
したがって、長期的な平均動脈圧レベルを新しい値に変更することは、塩と水の摂取量のレベルまたは腎機能曲線の圧力軸に沿ったシフトの程度のいずれか、または両方を変更しない限り不可能です。しかし、これらのいずれかが変更された場合、動脈圧はその後、2つの新しい曲線が交差する新しい圧力レベルで調節されることがわかります。
しかし、多くの人々では、腎機能曲線は図19-3に示されているものよりもはるかに急峻であり、塩の摂取量の変化が動脈圧に与える影響はわずかであると、次の節で説明します。
慢性的な腎排出曲線は急性曲線よりもはるかに急峻
圧ナトリウム利尿(pressure natriuresis)および圧利尿(pressure diuresis)の重要な特徴は、数日から数ヶ月続く動脈圧の慢性的な変化が、圧力の急性変化(図19-4)に比べて塩と水の腎排出に対してはるかに大きな影響を持つことです。したがって、腎臓が正常に機能している場合、慢性的な腎排出曲線は急性曲線よりもはるかに急峻です。
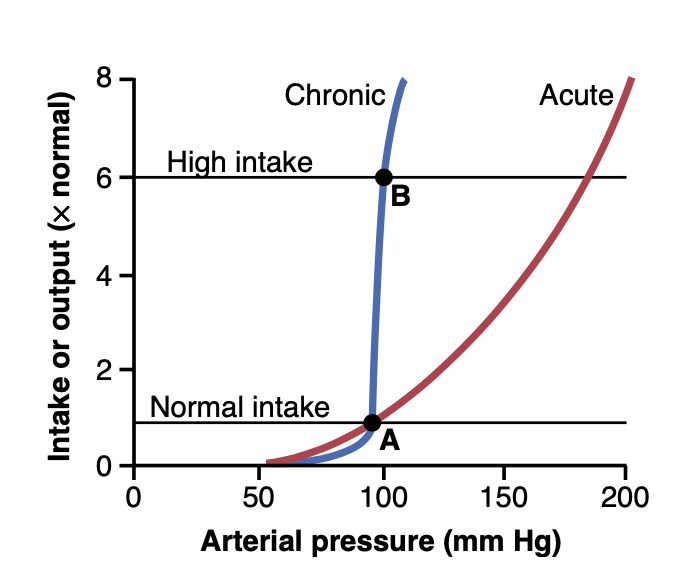
Figure 19-4. Acute and chronic renal output curves. Under steadystate conditions, the renal output of salt and water is equal to intake of salt and water. Points A and B represent the equilibrium points for long-term regulation of arterial pressure when salt intake is normal or six times normal, respectively. Because of the steepness of the chronic renal output curve, increased salt intake normally causes only small changes in arterial pressure. In persons with impaired kidney func- tion, the steepness of the renal output curve may be reduced, similar to the acute curve, resulting in increased sensitivity of arterial pres- sure to changes in salt intake.
慢性的な動脈圧上昇が尿排出に与える強力な影響は、圧力の上昇が腎臓に直接的な血行力学的(hemodynamic)効果をもたらし排泄を増加させるだけでなく、血圧上昇時に起こる神経およびホルモンの変化による間接的な効果も持っているためです。例えば、動脈圧の上昇は交感神経系(sympathetic nervous system)の活動を低下させ、部分的には第18章で説明した圧受容器反射機構(baroreceptor reflex mechanisms)を通じて行われます。また、アンジオテンシンII(angiotensin II)やアルドステロン(aldosterone)などの腎臓による塩と水の排泄を抑えるホルモンの生成を減少させます。これらの抗ナトリウム利尿系(antinatriuretic systems)の活動が低下するため、慢性的な動脈圧上昇時に塩と水の排泄を増加させる圧ナトリウム利尿および圧利尿の効果が強化されます(詳細は第28章および第30章を参照してください)。
逆に、血圧が低下すると交感神経系が活性化し、抗ナトリウム利尿ホルモンの生成が増加し、圧力低下による直接的な影響に加えて塩と水の腎排出を減少させます。腎臓に対する圧力の直接的な効果と交感神経系およびさまざまなホルモン系への圧力の間接的な効果の組み合わせにより、圧ナトリウム利尿および圧利尿は動脈圧と体液量の長期的な制御において非常に強力な要因となります。
ナトリウム摂取の慢性的な変化における圧ナトリウム利尿に対する神経およびホルモンの影響の重要性は特に顕著です。腎臓および神経およびホルモンの機構が正常に機能している場合、塩と水の摂取が通常の6倍に増加しても動脈圧に与える影響はほとんどありません。図中の血圧平衡点Bは、通常の塩摂取時の平衡点Aとほぼ同じであることに注目してください。逆に、塩と水の摂取量が通常の6分の1まで減少しても、通常は動脈圧にほとんど影響を与えません。このように、多くの人々は「塩感受性が低い(salt-insensitive)」とされ、塩摂取量の大きな変動でも血圧が数mmHg以上変化しないことがあります。
しかし、腎障害やアンジオテンシンIIやアルドステロンの過剰分泌を伴う人々は「塩感受性が高い(salt-sensitive)」ことがあり、図19-4に示されている急性曲線に似た減衰した腎排出曲線を持っています。このような場合、塩の摂取が中程度増加するだけで動脈圧が大幅に上昇することがあります。
血圧を塩感受性にする要因のいくつかには、腎障害による機能的なネフロンの喪失やアンジオテンシンIIやアルドステロンのような抗ナトリウム利尿ホルモンの過剰生成が含まれます。例えば、腎臓の外科的切除や高血圧、糖尿病、さまざまな腎疾患による腎損傷などは、塩摂取の変化に対して血圧をより感受性にします。このような場合、塩と水の摂取と排泄のバランスを維持するためには、腎排出を十分に増加させるために通常よりも高い動脈圧が必要です。
数年間続く長期的な高塩摂取が実際に腎臓を損傷し、最終的に血圧を塩感受性にする可能性があるという証拠もあります。本章の後半で高血圧患者における血圧の塩感受性について説明します。
体液摂取(fluid intake)と腎機能(renal function)が変化しない場合、全末梢抵抗(total peripheral resistance)の増加が動脈圧(arterial pressure)の長期レベルを上昇させない
動脈圧の基本的な方程式—動脈圧は心拍出量(cardiac output)と全末梢抵抗の積である—を思い出すと、全末梢抵抗が増加すれば動脈圧が上昇するはずです。確かに、全末梢抵抗が急激に増加すると動脈圧は即座に上昇します。しかし、腎臓が正常に機能し続けるならば、急性の動脈圧の上昇は通常維持されません。その代わり、動脈圧は1~2日以内に全て正常に戻ります。なぜでしょうか?
この現象の理由は、腎臓以外の体のすべての部位で血管抵抗(vascular resistance)が増加しても、腎臓によって決定される血圧制御の平衡点は変わらないからです(図19-1および図19-3を参照)。その代わり、腎臓は高い動脈圧に直ちに反応し、圧利尿(pressure diuresis)と圧ナトリウム利尿(pressure natriuresis)を引き起こします。数時間以内に大量の塩と水が体から失われ、この過程は動脈圧が平衡圧力レベルに戻るまで続きます。この時点で、動脈圧は正常化し、細胞外液量(extracellular fluid volume)および血液量は通常よりも低いレベルに減少します。
図19-5は、長期的な全末梢抵抗が通常よりもはるかに少ない、またははるかに大きい異なる臨床状態における心拍出量および動脈圧を示していますが、腎臓による塩と水の排泄は正常です。これらの異なる臨床状態すべてにおいて動脈圧も正常であることに注目してください。
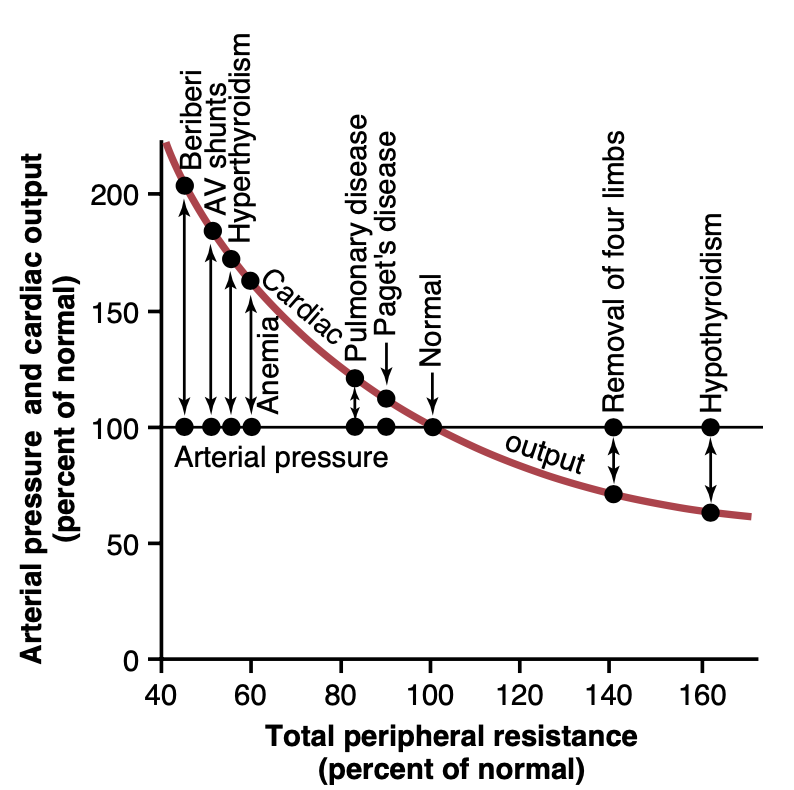
Figure 19-5. Relationships of total peripheral resistance to the long- term levels of arterial pressure and cardiac output in different clinical abnormalities. In these conditions, the kidneys were functioning nor- mally. Note that changing the whole-body total peripheral resistance caused equal and opposite changes in cardiac output but, in all cases, had no effect on arterial pressure. AV, Arteriovenous. (Modified from Guyton AC: Arterial Pressure and Hypertension. Philadelphia: WB Saunders, 1980.)
ここで注意が必要です。しばしば、全末梢抵抗が増加すると、腎内血管抵抗(intrarenal vascular resistance)も同時に増加し、腎臓の機能が変化して腎機能曲線(renal function curve)が高い圧力レベルにシフトし(図19-3A参照)、高血圧(hypertension)を引き起こすことがあります。この章の後半で血管収縮機構(vasoconstrictor mechanisms)による高血圧について説明する際にこのメカニズムの例を見ていきます。しかし、原因は腎抵抗の増加であり、全末梢抵抗の増加ではないことが重要な違いです。
体液量(fluid volume)の増加は心拍出量(cardiac output)または全末梢抵抗(total peripheral resistance)を増加させることで動脈圧(arterial pressure)を上昇させる
血管容量(vascular capacity)が同時に増加しない場合、細胞外液量(extracellular fluid volume)の増加が動脈圧を上昇させる全体的なメカニズムは図19-6に示されています。連続するイベントは次の通りです。
(1) 細胞外液量の増加、
(2) 血液量の増加、
(3) 平均循環充填圧(mean circulatory filling pressure)の増加、
(4) 心臓への静脈還流(venous return)の増加、
(5) 心拍出量の増加、
(6) 動脈圧の増加。
この増加した動脈圧は、腎臓の塩と水の排泄を増加させ、腎機能が正常で血管容量が変化していない場合、細胞外液量をほぼ正常に戻す可能性があります。
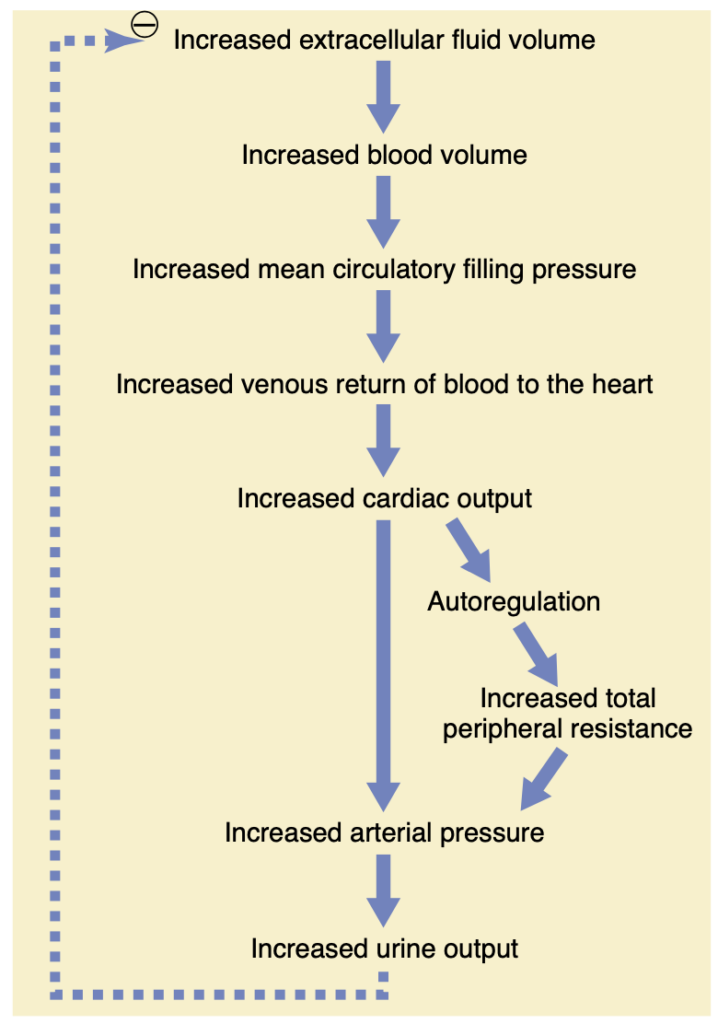
Figure 19-6. Sequential steps whereby increased extracellular fluid volume increases the arterial pressure. Note especially that increased cardiac output has both a direct effect to increase arterial pressure and an indirect effect by first increasing the total peripheral resistance.
この場合、特に注目すべきは、心拍出量(cardiac output)の増加が動脈圧(arterial pressure)を増加させる2つの方法です。1つは、心拍出量の直接的な増加によって圧力が増加する直接効果です。もう1つは、血流の自己調節(autoregulation)を通じて全末梢血管抵抗(total peripheral resistance)を上昇させる間接的な効果です。この2つ目の効果について説明します。
第17章に関連して、組織を流れる血液の量が過剰になると、局所的な組織の血管が収縮し、血流を正常に戻すという現象があることを思い出してください。これを血流の自己調節(autoregulation)と呼び、組織自体による血流の調節を意味します。血液量の増加が心拍出量を上昇させると、体のすべての組織で血流が増加する傾向があります。もしこの増加した血流が組織の代謝ニーズを超える場合、自己調節メカニズムが体中の血管を収縮させ、その結果として全末梢抵抗が増加します。
最後に、動脈圧が心拍出量と全末梢抵抗の積であるため、自己調節メカニズムによる全末梢抵抗の二次的な増加が動脈圧の上昇を助けます。例えば、心拍出量のわずか5〜10%の増加でも、血流の自己調節や血管収縮を引き起こす他の要因による全末梢抵抗の増加を伴うと、動脈圧を通常の平均動脈圧100 mmHgから150 mmHgまで上昇させることができます。このわずかな心拍出量の増加は、しばしば測定不可能なことが多いです。
動脈圧(arterial pressure)調節における腎・体液スキーマにおける塩(NaCl)の重要性
これまでの議論では、動脈圧の調節における体液量の重要性が強調されてきましたが、実験的な研究では、塩摂取の増加は水摂取の増加よりも動脈圧を上昇させる可能性が高いことが示されています。特に塩感受性(salt-sensitive)のある人々においてはその傾向が顕著です。この理由は、純粋な水は通常、摂取されるとすぐに腎臓から排出されるのに対し、塩はそう簡単には排出されないためです。塩が体内に蓄積すると、以下の2つの基本的な理由から細胞外液量(extracellular fluid volume)も間接的に増加します。
- 塩が体内に蓄積すると、いくらかのナトリウム(sodium)が組織に蓄積されることはありますが、細胞外液中の過剰な塩分が体液の浸透圧(osmolality)を増加させます。この浸透圧の増加は脳の渇き中枢(thirst center)を刺激し、体は余分な水を飲むことで細胞外の塩濃度を正常に戻し、細胞外液量を増加させます。
- 細胞外液中の過剰な塩による浸透圧の増加は、視床下部–後部下垂体腺(hypothalamic–posterior pituitary gland)の分泌メカニズムを刺激し、抗利尿ホルモン(antidiuretic hormone)の分泌を増加させます(第29章で説明)。抗利尿ホルモンは腎臓が腎尿細管液(renal tubular fluid)から大量の水を再吸収するように働き、尿の排泄量を減少させ、細胞外液量を増加させます。
したがって、体内に蓄積される塩の量は細胞外液量の重要な決定因子です。比較的小さな細胞外液および血液量の増加でも動脈圧を大幅に増加させることがあります。しかし、これは過剰な塩の蓄積が血液量の増加を引き起こし、かつ血管容量が同時に増加しない場合に限ります。前述のように、腎機能に障害がないか、抗ナトリウム利尿ホルモン(antinatriuretic hormones)の過剰形成がない限り、塩摂取の増加は通常、動脈圧をあまり増加させません。これは、腎臓が余分な塩を迅速に排出し、血液量がほとんど変化しないためです。
腎機能障害による慢性高血圧(CHRONIC HYPERTENSION)
人が慢性高血圧(chronic hypertension)または高血圧(high blood pressure)と診断されるとき、それはその人の平均動脈圧(mean arterial pressure)が受け入れられている正常範囲の上限を超えていることを意味します。平均動脈圧が110 mmHg(正常は約90 mmHg)を超える場合、高血圧と見なされます。(この平均圧レベルは、拡張期血圧(diastolic blood pressure)が約90 mmHgを超え、収縮期圧(systolic pressure)が約135 mmHgを超えるときに発生します。)重度の高血圧のある人では、平均動脈圧が150~170 mmHgに達し、拡張期圧は最大で130 mmHg、収縮期圧は時折250 mmHgに達することがあります。
動脈圧が中程度に上昇しても、寿命は短くなります。重度の高圧、つまり平均動脈圧が通常の50%以上上昇している場合、適切な治療を受けない限り、その人は数年以内にしか生きられません。高血圧の致命的な影響は主に以下の3つの方法で引き起こされます。
- 心臓への過剰な負荷が早期の心不全(heart failure)および冠動脈性心疾患(coronary heart disease)を引き起こし、しばしば心臓発作(heart attack)による死をもたらします。
- 高血圧はしばしば脳内の主要な血管を損傷し、その後脳の主要部分が死滅します。この現象は脳梗塞(cerebral infarct)と呼ばれ、臨床的には脳卒中(stroke)と呼ばれます。どの脳の部分が関与しているかにより、脳卒中は致命的であったり、麻痺(paralysis)、認知症(dementia)、失明(blindness)、またはその他の深刻な脳障害を引き起こす可能性があります。
- 高血圧はほぼ常に腎臓に損傷を引き起こし、多くの腎破壊(renal destruction)領域を生じ、最終的には腎不全(kidney failure)、尿毒症(uremia)、そして死に至ります。
「容量負荷性高血圧(volume-loading hypertension)」と呼ばれるタイプの高血圧から得られた教訓は、動脈圧調節における腎・体液量機構(renal–body fluid volume mechanism)の役割を理解する上で重要です。容量負荷性高血圧とは、体内の細胞外液量の過剰な蓄積によって引き起こされる高血圧を指し、いくつかの例が以下に示されます。
腎質量減少と塩摂取増加による実験的容量負荷性高血圧(volume-loading hypertension)
図19-7は、腎質量(kidney mass)の70%を切除した犬のグループにおける容量負荷性高血圧を示す典型的な実験を示しています。曲線上の最初の円で囲まれた点では、一方の腎臓の両極が除去され、次の円で囲まれた点では反対側の腎臓全体が除去され、動物は正常な腎質量の30%のみが残されました。この腎質量の除去により、動脈圧が平均で6 mmHgだけ上昇しました。その後、犬には水の代わりに塩溶液を飲ませました。塩溶液は喉の渇きを癒すことができないため、犬は通常の2倍から4倍の量を飲み、数日以内に平均動脈圧が通常より約40 mmHg上昇しました。2週間後、犬には再び水道水が与えられ、圧力は2日以内に正常に戻りました。最後に実験の終わりに再び塩溶液を与えたところ、今回は圧力がはるかに迅速に高レベルまで上昇し、再び容量負荷性高血圧が示されました。
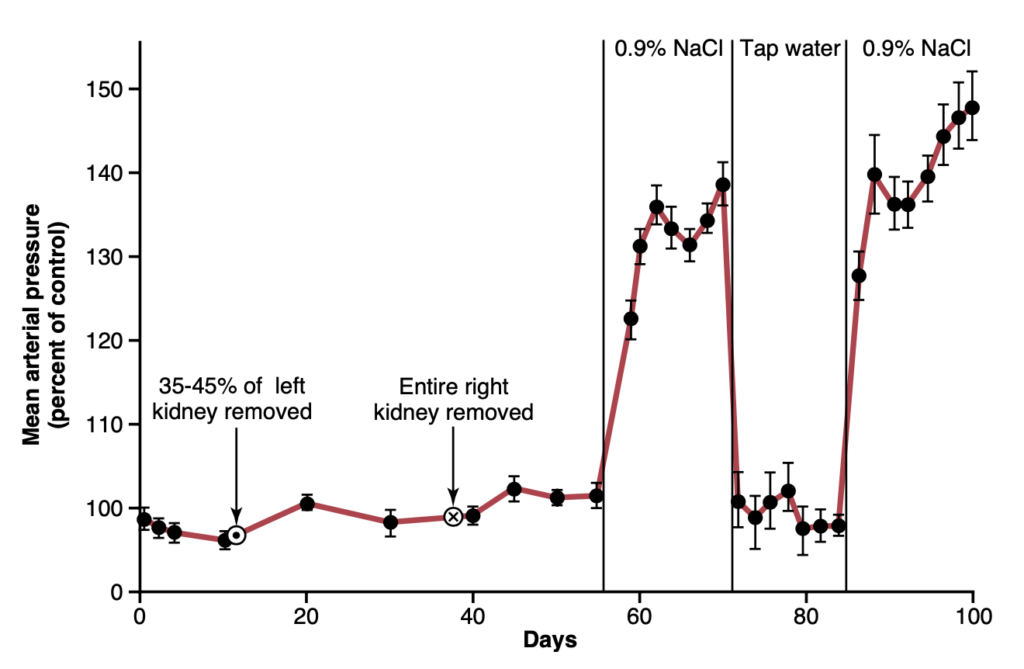
Figure 19-7. The average effect on ar- terial pressure of drinking 0.9% saline solution (0.9% NaCl) instead of water in dogs with 70% of their renal tissue removed. (Modified from Langston JB, Guyton AC, Douglas BH, et al: Effect of changes in salt intake on arterial pressure and renal function in partially nephrectomized dogs. Circ Res 12:508, 1963.)
図19-7に示された容量負荷実験において高血圧が発生した理由を、長期的な動脈圧調節の基本的な決定因子を再度考慮すると理解できます。まず、腎質量を正常の30%に減少させることで、腎臓が塩と水を排泄する能力が大幅に低下しました。したがって、塩と水が体内に蓄積し、数日以内に動脈圧が上昇して余分な塩と水の摂取を排泄するに十分なレベルに達しました。
容量負荷性高血圧の発達中の循環機能の連続的変化
容量負荷性高血圧(volume-loading hypertension)の進行中の循環機能の連続的変化(図19-8)を研究することは特に有益です。「0」日と表示される数週間前に、腎質量はすでに正常の30%に減少していました。そして、この時点で塩と水の摂取量が通常の約6倍に増加され、その後も高摂取が続けられました。急性の影響は、細胞外液量(extracellular fluid volume)、血液量(blood volume)、および心拍出量(cardiac output)を20%から40%増加させることでした。同時に動脈圧も上昇し始めましたが、最初は体液量や心拍出量ほど大きくは上昇しませんでした。この圧力の上昇が遅い理由は、全末梢抵抗(total peripheral resistance)曲線を研究することで理解できます。この曲線は全末梢抵抗が初期に減少したことを示しており、第18章で説明した圧受容器機構(baroreceptor mechanism)によって圧力の上昇が一時的に抑制されたためです。しかし、2~4日後には圧受容器が適応(リセット)し、圧力の上昇を防ぐことができなくなりました。この時点で、全末梢抵抗がほぼ正常なレベルであっても、心拍出量の増加により動脈圧はほぼ最大まで上昇しました。
Figure 19-8. Progressive changes in important circulatory system variables during the first few weeks of volume-loading hypertension. Note especially the initial increase in cardiac output as the basic cause of the hypertension. Subsequently, the autoregulation mechanism returns the cardiac output almost to normal while simultaneously causing a secondary increase in total peripheral resistance. (Modified from Guyton AC: Arterial Pressure and Hypertension. Philadelphia: WB Saunders, 1980.)
これらの早期の急性変化が起こった後、次の数週間の間により長期的な二次変化が発生しました。特に重要だったのは全末梢抵抗の進行的な増加であり、同時に心拍出量が正常に戻る方向に減少しました。この減少は少なくとも部分的には、第17章および本章で前述した長期的な血流自己調節機構(long-term blood flow autoregulation mechanism)の結果でした。すなわち、心拍出量が高レベルに上昇し高血圧を引き起こした後、組織を通る過剰な血流が局所細動脈の進行的な収縮を引き起こし、体組織の局所血流と心拍出量を正常に戻す一方で、全末梢抵抗の二次的な増加を引き起こしました。
細胞外液量と血液量も心拍出量の減少とともに正常に戻ったことに注目してください。この結果は2つの要因によるものでした。第一に、細動脈抵抗の増加により毛細血管圧が低下し、組織空間内の体液が血液に再吸収されることが可能になりました。第二に、上昇した動脈圧により、腎臓が最初に体内に蓄積した余分な体液を排泄することになりました。
容量負荷の初期発症から数週間後、以下の影響が見られました。
- 高血圧(hypertension)
- 全末梢抵抗の著しい増加
- 細胞外液量、血液量、および心拍出量のほぼ完全な正常化
したがって、容量負荷性高血圧を2つの連続した段階に分けることができます。第一段階は、体液量の増加による心拍出量の増加から生じるもので、この心拍出量の増加が高血圧を媒介します。容量負荷性高血圧の第二段階は、高血圧および高い全末梢抵抗が特徴であり、心拍出量は正常に非常に近いレベルまで戻り、通常の測定技術では異常に高い心拍出量を検出することができないことがしばしばあります。
したがって、容量負荷性高血圧における全末梢抵抗の増加は、高血圧が発生した後に生じるものであり、したがって高血圧の原因ではなく、その結果であることが分かります。
腎臓がなく人工腎(artificial kidney)で維持されている患者における容量負荷性高血圧(volume-loading hypertension)
患者が人工腎で維持されている場合、透析(dialysis)のたびに適切な量の水と塩を除去して体液量を正常に保つことが特に重要です。このステップが行われず、細胞外液量(extracellular fluid volume)が増加すると、図19-8に示されるのと全く同じ方法で高血圧(hypertension)がほぼ必然的に発生します。すなわち、まず心拍出量(cardiac output)が増加し、高血圧を引き起こします。そして、自己調節(autoregulation)メカニズムにより心拍出量が正常に戻る一方で、全末梢抵抗(total peripheral resistance)が二次的に増加します。したがって、最終的にこの高血圧は高末梢抵抗型の高血圧のように見えますが、初期の原因は体液の過剰蓄積です。
過剰なアルドステロン(aldosterone)による高血圧
もう一つの容量負荷性高血圧のタイプは、体内に過剰なアルドステロンやその他のステロイドが存在することによって引き起こされます。副腎(adrenal gland)の小さな腫瘍が大量のアルドステロンを分泌することがあり、この状態を原発性アルドステロン症(primary aldosteronism)と呼びます(第28章および第30章で説明)。アルドステロンは腎臓の尿細管(tubules)による塩と水の再吸収速度を増加させるため、血液量、細胞外液量、および動脈圧が増加します。塩摂取が同時に増加すると、高血圧はさらに大きくなります。さらに、この状態が数ヶ月または数年続くと、過剰な動脈圧が腎臓に病理学的な変化を引き起こし、腎臓がアルドステロンによる直接的な影響に加えてさらに多くの塩と水を保持するようになります。そのため、高血圧は最終的に致命的なレベルに達することが多いです。
このタイプの高血圧の初期段階でも、心拍出量はしばしば増加しますが、後の段階では心拍出量はほぼ正常に戻り、全末梢抵抗が二次的に増加することが一般的です。これは本章の初めに説明した原発性容量負荷性高血圧(primary volume-loading hypertension)と同様です。
動脈圧(arterial pressure)制御におけるレニン–アンジオテンシン系(renin-angiotensin system)の役割 ※これはマスタしなといけない!
腎臓は細胞外液量の変化を通じて動脈圧を制御する能力を持つほかに、圧力を制御するためのもう一つの強力なメカニズム、すなわちレニン–アンジオテンシン系を持っています。
レニン(renin)は、動脈圧が低すぎると腎臓から放出されるタンパク質酵素であり、それによって動脈圧がいくつかの方法で上昇し、初期の圧力低下を修正するのに役立ちます。
レニン–アンジオテンシン系の構成要素
図19-9は、レニン–アンジオテンシン系が動脈圧を調節するのを助ける主な機能的ステップを示しています。レニンは腎臓の傍糸球体細胞(juxtaglomerular cells、JG細胞)で合成・貯蔵されています。JG細胞は、主に糸球体(glomeruli)に近い輸入細動脈(afferent arterioles)の壁に位置する修飾された平滑筋細胞です。レニン分泌を制御する要因には、交感神経系(sympathetic nervous system)、さまざまなホルモン、およびプロスタグランジン(prostaglandins)、一酸化窒素(nitric oxide)、エンドセリン(endothelin)などの局所的なオータコイド(autacoid)が含まれます。動脈圧が低下すると、JG細胞は少なくとも以下の3つの主要なメカニズムでレニンを分泌します。
- JG細胞の圧感受性圧受容器(pressure-sensitive baroreceptors)が動脈圧の低下に反応してレニンの分泌を増加させます。
- 初期遠位尿細管(early distal tubule)の緻密斑細胞(macula densa cells)への塩化ナトリウム(sodium chloride)供給の減少がレニンの分泌を刺激します(第27章でさらに詳しく説明します)。
- 交感神経系活動の増加が、JG細胞のベータアドレナリン受容体(beta-adrenergic receptors)を活性化することでレニンの分泌を刺激します。交感神経刺激はまた、アルファアドレナリン受容体(alpha-adrenergic receptors)を活性化し、強い交感神経活動の場合に腎臓の塩化ナトリウム再吸収を増加させ、糸球体濾過率(glomerular filtration rate)を減少させます。腎交感神経活動の増加は、レニン分泌のための腎圧受容器および緻密斑メカニズムの感受性を高めることもあります。
レニンのほとんどは腎血流に入り、腎臓を通り抜けて全身に循環します。ただし、少量のレニンは腎臓の局所体液に残り、いくつかの腎内機能を開始します。
レニン自体は酵素であり、血管活性物質(vasoactive substance)ではありません。図19-9に示されているように、レニンは別の血漿タンパク質であるレニン基質(renin substrate、またはアンジオテンシノーゲン(angiotensinogen))に酵素的に作用し、10アミノ酸ペプチドであるアンジオテンシンI(angiotensin I)を放出します。アンジオテンシンIは軽度の血管収縮作用を持っていますが、循環機能に大きな変化を引き起こすほどの力はありません。レニンは血液中に30~60分間持続し、その間にさらに多くのアンジオテンシンIを形成し続けます。
Figure 19-9. The renin-angiotensin vasoconstrictor mechanism for arterial pressure control.
アンジオテンシンIが形成されてから数秒から数分以内に、さらに2つのアミノ酸がアンジオテンシンIから切り離され、8アミノ酸ペプチドであるアンジオテンシンII(angiotensin II)が形成されます。この変換は、血液が肺の小血管を通る際に肺血管の内皮(endothelium)に存在するアンジオテンシン変換酵素(angiotensin-converting enzyme、ACE)によって大部分が行われます。他の組織(腎臓や血管など)もACEを含んでいるため、局所的にアンジオテンシンIIを形成します。
アンジオテンシンIIは非常に強力な血管収縮物質であり、循環機能にも他の方法で影響を与えますが、血液中にはわずか1〜2分しか持続しません。これは、アンジオテンシナーゼ(angiotensinases)と呼ばれる多くの血液および組織酵素によって迅速に不活性化されるためです。
アンジオテンシンIIには動脈圧を上昇させる2つの主要な効果があります。
1. 最初の効果は、体内の多くの部位で血管収縮(vasoconstriction)を引き起こすことで、これは迅速に起こります。動脈(arterioles)で強く収縮し、静脈(veins)ではやや少ない収縮を引き起こします。動脈の収縮は全末梢抵抗を増加させ、それによって動脈圧を上昇させます(図19-9の下部に示されています)。
2. また、静脈の軽度の収縮は心臓への静脈還流(venous return)を増加させ、心臓が増加した圧力に対してポンプするのを助けます。
アンジオテンシンIIが動脈圧を上昇させる第2の主要な手段は、アルドステロン分泌の刺激や腎臓への直接的な効果による腎臓での塩と水の排泄の減少です。腎臓による塩と水の保持は細胞外液量をゆっくりと増加させ、その後数時間から数日にわたって動脈圧を増加させます。長期的な効果として、アンジオテンシンIIの腎臓に対する直接および間接的な作用は、最終的に動脈圧を上昇させる急性の血管収縮メカニズムよりもさらに強力です。
レニン–アンジオテンシン系の圧受容器圧反応の迅速さと強さ
図19-10は、出血(hemorrhage)が動脈圧に与える影響を示す実験の結果を示しており、2つの条件下で行われました。(1) レニン–アンジオテンシン系が機能している場合、および (2) レニンをブロックする抗体でシステムをブロックした場合です。出血後、動脈圧が急激に50 mmHgまで低下した場合、レニン–アンジオテンシン系が機能していると動脈圧は83 mmHgまで回復しました。
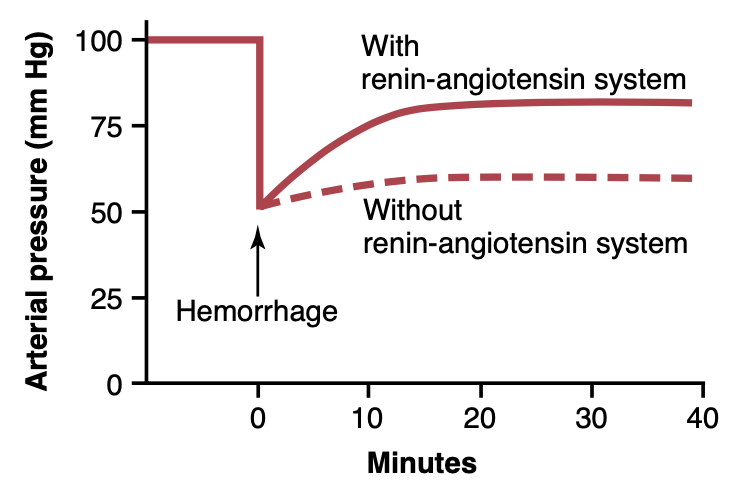
Figure 19-10. The pressure-compensating effect of the renin- angiotensin vasoconstrictor system after severe hemorrhage. (Drawn from experiments by Dr. Royce Brough.)
一方、レニン–アンジオテンシン系がブロックされた場合、動脈圧は60 mmHgまでしか回復しませんでした。この現象は、レニン–アンジオテンシン系が、重度の出血後、動脈圧を数分以内に少なくとも半分程度正常に戻すほど強力であることを示しています。したがって、このシステムは特に循環ショック(circulatory shock)の際に体にとって生命を救う役割を果たすことができます。
また、レニン–アンジオテンシン血管収縮系が完全に活性化されるには約20分かかることにも注意してください。したがって、このシステムは血圧制御のためには神経反射や交感神経のノルエピネフリン–エピネフリン系(norepinephrine-epinephrine system)よりもやや遅いです。
アンジオテンシンIIは塩と水の腎保持を引き起こす—動脈圧(arterial pressure)の長期制御のための重要な手段
アンジオテンシンIIは、腎臓での塩と水の保持を2つの主要な方法で引き起こします。
- アンジオテンシンIIは腎臓に直接作用して塩と水の保持を引き起こします。
- アンジオテンシンIIは副腎(adrenal glands)を刺激してアルドステロン(aldosterone)を分泌させ、そのアルドステロンが腎尿細管による塩と水の再吸収を増加させます。
したがって、アンジオテンシンIIが血中に過剰に循環すると、動脈圧制御のための長期的な腎・体液メカニズム全体が通常より高い動脈圧レベルに自動的に設定されます。
アンジオテンシンIIの塩と水の腎保持を引き起こす直接的な腎作用のメカニズム
アンジオテンシンIIは腎臓に対して塩と水の保持を促進するいくつかの直接的な作用を持っています。主な効果の一つは、腎動脈(renal arterioles)、特に糸球体の輸出細動脈(glomerular efferent arterioles)を収縮させることで、腎臓の血流を減少させます。血流がゆっくりになると、尿細管周囲毛細血管(peritubular capillaries)の圧力が低下し、尿細管からの体液の再吸収が増加します。さらに、アンジオテンシンIIは尿細管細胞(tubular cells)に直接作用してナトリウムと水の再吸収を増加させる重要な作用を持っています(第28章で説明)。アンジオテンシンIIのこれらの複合的な効果により、尿の排出量は通常の5分の1以下に減少することもあります。
アンジオテンシンIIはアルドステロン分泌を刺激して腎臓での塩と水の保持を増加させる
アンジオテンシンIIは、副腎によるアルドステロンの分泌を最も強力に刺激する物質の一つでもあります。これは第30章で体液の調節に関して、第78章で副腎機能に関してさらに詳しく説明します。したがって、レニン–アンジオテンシン系が活性化されると、アルドステロン分泌率も通常増加します。アルドステロンの重要な後続の機能は、腎尿細管によるナトリウム再吸収を著しく増加させることであり、これにより体全体の細胞外液ナトリウムおよび細胞外液量が増加します。このように、アンジオテンシンIIの腎臓に対する直接的な効果とアルドステロンを介した効果の両方が、長期的な動脈圧制御において重要です。
アンジオテンシンIIによる動脈圧(arterial pressure)の変化の定量的分析
図19-11は、動脈圧制御におけるアンジオテンシンの影響を定量的に分析したものです。この図には2つの腎機能曲線(renal function curves)と、ナトリウム摂取の正常レベルを示す線が描かれています。左側の腎機能曲線は、アンジオテンシンIからアンジオテンシンIIへの変換を阻害するACE阻害薬を使用してレニン–アンジオテンシン系をブロックした犬で測定されたものです。右側の曲線は、アンジオテンシンIIを通常の血中形成速度の約2.5倍のレベルで連続的に投与された犬で測定されたものです。アンジオテンシンIIの影響下で腎排出曲線が高い圧力レベルにシフトしていることに注目してください。このシフトは、腎臓に対するアンジオテンシンIIの直接的な効果とアルドステロン(aldosterone)分泌を通じた間接的な効果によって引き起こされます。
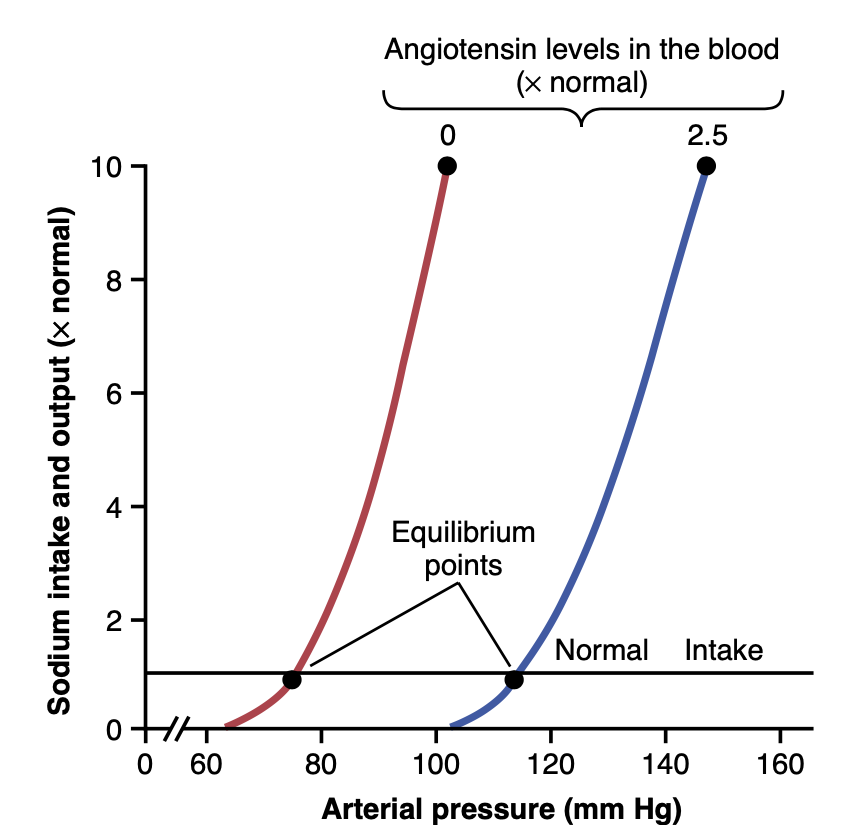
Figure 19-11. The effect of two angiotensin II levels in the blood on the renal output curve showing regulation of the arterial pressure at an equilibrium point of 75 mm Hg, when the angiotensin II level is low, and at 115 mm Hg, when the angiotensin II level is high.
最後に、アンジオテンシンがゼロの場合に75 mmHgの動脈圧レベルを示す平衡点と、アンジオテンシンが増加した場合に115 mmHgの圧力レベルを示す平衡点の2つの平衡点に注目してください。したがって、アンジオテンシンによる塩と水の腎保持は、動脈圧の慢性的な上昇を促進する上で強力な効果を持つ可能性があります。
レニン–アンジオテンシン系が塩摂取量の大きな変動にもかかわらず正常な動脈圧を維持する役割
レニン–アンジオテンシン系の最も重要な機能の一つは、非常に少ない量または非常に多量の塩を摂取しても、細胞外液量(extracellular fluid volume)や動脈圧に大きな変化を引き起こさないようにすることです。この機能は図19-12で説明されています。塩摂取の増加の初期効果は細胞外液量を増加させ、それが動脈圧を上昇させる傾向にあります。塩摂取の増加による多くの効果、特に動脈圧のわずかな上昇や圧力に依存しない効果により、レニン分泌およびアンジオテンシンII形成の速度が減少し、それにより細胞外液量または動脈圧のわずかな増加で余分な塩を排除することができます。したがって、レニン–アンジオテンシン系は、塩摂取が増加しても動脈圧を正常または正常に近いレベルに維持する自動フィードバック機構です。塩摂取が正常以下に減少すると、全く逆の効果が起こります。
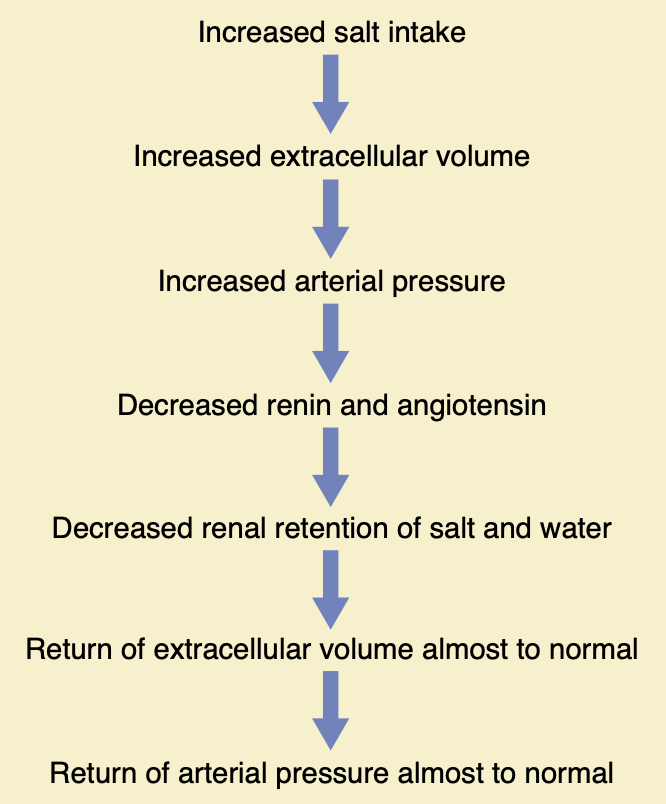
Figure 19-12. Sequential events whereby increased salt intake in- creases the arterial pressure, but feedback decrease in activity of the renin angiotensin system returns the arterial pressure almost to the normal level.
レニン–アンジオテンシン系が動脈圧を制御する効力を強調するために、このシステムが正常に機能している場合、塩摂取が100倍に増加しても動脈圧は通常4〜6 mmHgしか上昇しません(図19-13)。逆に、アンジオテンシンIIの形成の通常の抑制が小量のアンジオテンシンIIの連続投与によって妨げられ、血中濃度が減少できない場合、同じ塩摂取の増加が40 mmHg以上の圧力上昇を引き起こすことがあります(図19-13を参照)。塩摂取が通常の10分の1まで減少しても、レニン–アンジオテンシン系が正常に機能している限り動脈圧はほとんど変化しません。しかし、アンジオテンシンIIの形成がACE阻害薬でブロックされると、塩摂取が減少するにつれて血圧は著しく低下します(図19-13を参照)。したがって、レニン–アンジオテンシン系は、塩摂取量の大きな変動に対応しつつ動脈圧の変化を最小限に抑える体の最も強力なシステムであるといえます。
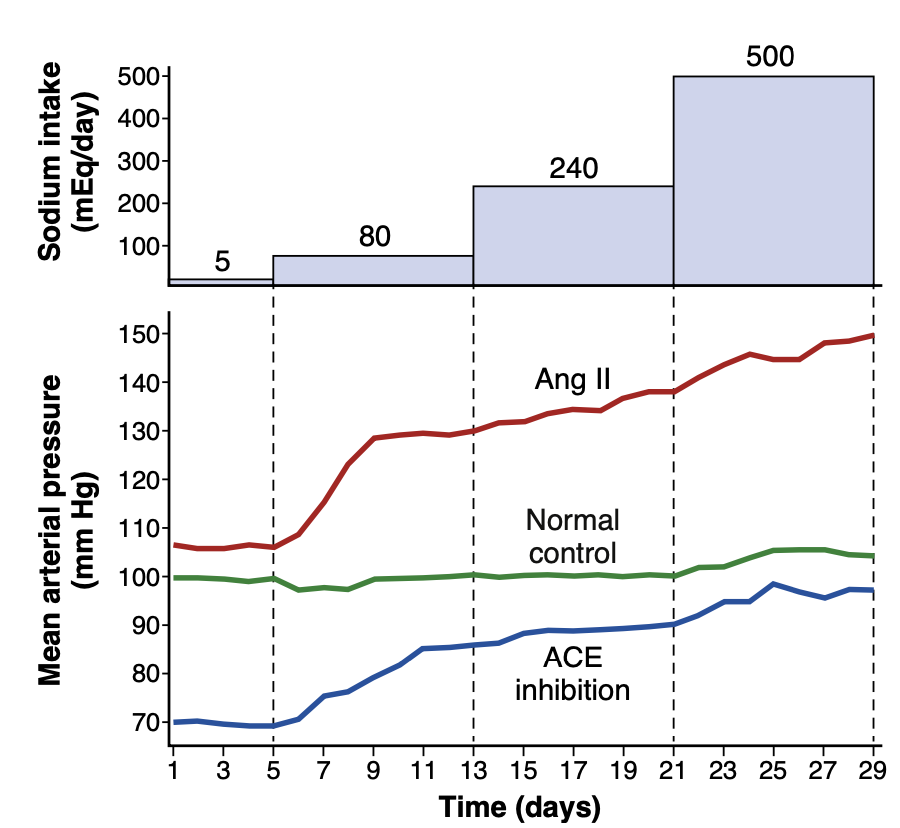
Figure 19-13. Changes in mean arterial pressure during chronic changes in sodium intake in normal control dogs and in dogs treated with an angiotensin-converting enzyme (ACE) inhibitor to block an- giotensin II (Ang II) formation or infused with Ang II to prevent Ang II from being suppressed. Sodium intake was raised in steps from a low level of 5 mmol/day to 80, 240, and 500 mmol/day for 8 days at each level. (Modified from Hall JE, Guyton AC, Smith MJ Jr, et al: Blood pressure and renal function during chronic changes in sodium intake: role of angiotensin. Am J Physiol 239:F271, 1980.)
レニン分泌腫瘍または腎虚血(renal ischemia)による高血圧
稀に、レニン分泌を行うJG細胞の腫瘍が発生し、大量のレニンを分泌して大量のアンジオテンシンIIを形成し、全患者において重度の高血圧が発生することがあります。また、大量のアンジオテンシンIIを数日または数週間にわたって連続的に動物に投与すると、同様に重度の長期高血圧が発生します。
アンジオテンシンIIが動脈圧を上昇させる方法についてはすでに述べましたが、それには2つの方法があります。
- 全身の細動脈(arterioles)を収縮させ、それによって全末梢抵抗(total peripheral resistance)および動脈圧を増加させること。この効果は大量のアンジオテンシンIIの投与を始めた直後から数秒以内に発生します。
- 腎臓での塩と水の保持を引き起こすこと。数日にわたって、適度な量のアンジオテンシンIIであっても、その腎作用によって高血圧を引き起こし、これが動脈圧の長期上昇の主な原因となります。
一腎ゴールドブラット高血圧(One-Kidney Goldblatt Hypertension)
一方の腎臓が除去され、残りの腎臓の腎動脈に収縮帯が装着されると(図19-14に示されているように)、直ちに収縮帯を越えた腎動脈の圧力が大幅に低下し、図の破線で示されています。その後、数秒から数分以内に全身の動脈圧が上昇し始め、数日間上昇し続けます。通常、最初の1時間ほどは圧力が急速に上昇し、その後数日間にわたってさらにゆっくりと上昇します。全身の動脈圧が新しい安定した圧力レベルに達すると、収縮部より下流の腎動脈圧(図の破線)はほぼ正常に戻ります。このようにして生じる高血圧を「一腎ゴールドブラット高血圧」と呼び、腎動脈収縮による高血圧の重要な定量的特徴を最初に研究したハリー・ゴールドブラットに敬意を表しています。
ゴールドブラット高血圧における動脈圧の初期上昇は、レニン–アンジオテンシン血管収縮機構によって引き起こされます。つまり、腎動脈が急性に収縮した後、腎臓への血流が悪いため、大量のレニンが腎臓から分泌され(図19-14の最下部の曲線で示されています)、これによりアンジオテンシンIIおよびアルドステロンのレベルが血中で増加します。アンジオテンシンIIは動脈圧を急性に上昇させます。レニンの分泌は約1〜2時間でピークに達し、5〜7日後にはほぼ正常に戻ります。この時点で腎動脈圧も正常に戻るため、腎臓はもはや虚血状態ではなくなります。
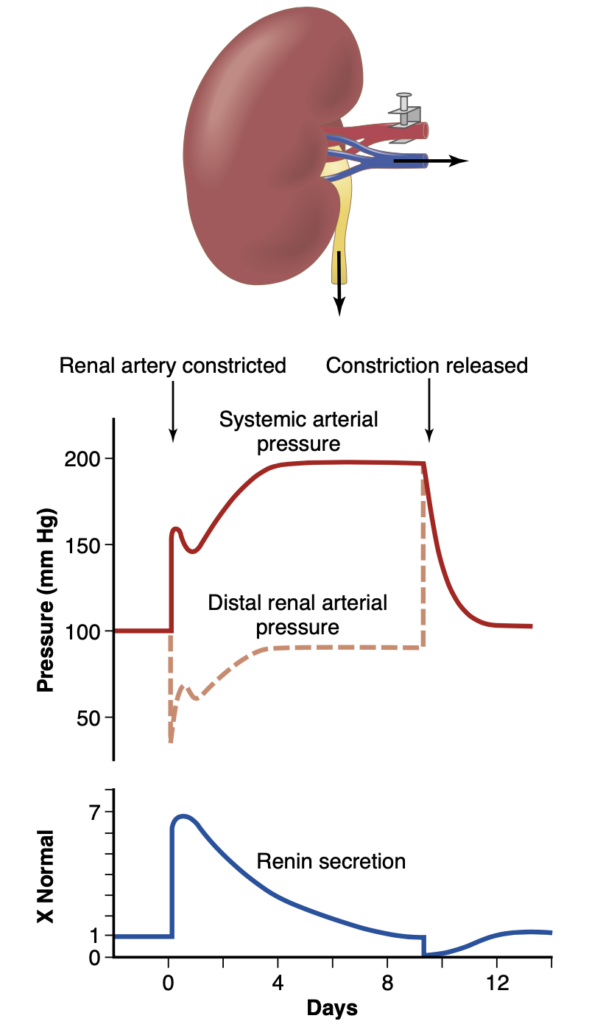
Figure 19-14. Effect of placing a constricting clamp on the renal artery of one kidney after the other kidney has been removed. Note the changes in systemic arterial pressure, renal artery pressure distal to the clamp, and rate of renin secretion. The resulting hypertension is called one-kidney Goldblatt hypertension.
動脈圧の2度目の上昇は、収縮した腎臓による塩と水の保持によって引き起こされ、これはアンジオテンシンIIとアルドステロンによっても刺激されます。5〜7日後、体液量が十分に増加し、動脈圧が新たな持続レベルに達します。この持続的な圧力レベルの定量値は、腎動脈の収縮の程度によって決まります。すなわち、収縮部より下流の腎動脈圧が正常な尿排出を引き起こすのに十分なレベルに達するように、大動脈圧は上昇する必要があります。
同様のシナリオが、単一の残存腎臓の腎動脈狭窄(stenosis)が発生した患者にも見られ、これは腎移植後に起こることがあります。また、動脈硬化(atherosclerosis)や過剰な血管収縮物質のレベルによる腎動脈の抵抗の機能的または病的な増加も、主な腎動脈の収縮と同じメカニズムで高血圧を引き起こすことがあります。
二腎ゴールドブラット高血圧(Two-Kidney Goldblatt Hypertension)
一方の腎臓の動脈が収縮し、他方の腎臓の動脈が正常な場合でも高血圧(hypertension)が発生することがあります。収縮した腎臓はレニン(renin)を分泌し、腎動脈圧の低下により塩と水を保持します。その後、反対側の「正常な」腎臓も虚血腎(ischemic kidney)によって生成されたレニンのために塩と水を保持します。このレニンはアンジオテンシンII(angiotensin II)およびアルドステロン(aldosterone)の形成を促進し、これらが反対側の腎臓に循環して塩と水を保持させます。このように、両方の腎臓が異なる理由で塩と水を保持するようになります。結果として、高血圧が発生します。
二腎ゴールドブラット高血圧の臨床的な対応例は、2つの腎臓を持つ人において動脈硬化(atherosclerosis)などで片方の腎動脈に狭窄が生じた場合に見られます。
レニンを慢性的に分泌する病変腎による高血圧
しばしば、1つまたは両方の腎臓の斑状領域が病変を持ち、局所的な血管収縮や梗塞(infarction)によって虚血状態になりますが、他の腎領域は正常です。この場合、ほぼ二腎タイプのゴールドブラット高血圧と同様の効果が発生します。すなわち、斑状の虚血腎組織がレニンを分泌し、これがアンジオテンシンIIを介して残りの腎質も塩と水を保持させます。腎性高血圧(renal hypertension)の最も一般的な原因の一つは、この斑状虚血腎疾患であり、特に高齢者に多く見られます。
容量負荷と血管収縮の組み合わせによるその他のタイプの高血圧
大動脈縮窄(Coarctation of the Aorta)による上半身の高血圧
数千人に1人の赤ちゃんが、大動脈が病的に収縮または閉塞した状態で生まれます。この収縮は、頭部と腕への大動脈動脈分枝の先で、腎動脈の手前で起こることがあります。この状態を「大動脈縮窄(coarctation of the aorta)」と呼びます。この場合、下半身への血流は体壁内の複数の小さな側副動脈によって運ばれ、上大動脈と下大動脈の間には多くの血管抵抗があります。その結果、上半身の動脈圧は下半身の動脈圧よりも40〜50%高くなることがあります。
この上半身の高血圧のメカニズムは、片腎ゴールドブラット高血圧(one-kidney Goldblatt hypertension)とほぼ同じです。すなわち、腎動脈の上流に収縮帯が置かれると、両腎の血圧がまず低下し、レニンが分泌され、アンジオテンシンおよびアルドステロンが形成され、上半身に高血圧が発生します。腎臓のレベルでの下半身の動脈圧はほぼ正常に上昇しますが、上半身では高圧が持続します。腎臓はもはや虚血状態ではないため、レニンの分泌およびアンジオテンシンとアルドステロンの形成はほぼ正常に戻ります。同様に、大動脈縮窄の場合、下半身の動脈圧は通常ほぼ正常であるのに対し、上半身の圧力は非常に高くなります。
大動脈縮窄による高血圧における自己調節の役割
大動脈縮窄による高血圧の重要な特徴は、圧力が通常より40〜60%高い腕における血流がほぼ正常であることです。また、圧力が上昇していない脚における血流もほぼ正常です。上半身の圧力が下半身よりも40〜60%高いにもかかわらず、なぜこのようなことが起こるのでしょうか。上半身と下半身の血液に含まれる血管収縮物質には違いはありません。なぜなら、同じ血液が両方の領域に流れているからです。同様に、神経系も両方の循環領域に同様に分布しているため、血管の神経制御に違いがあるとは考えられません。主な理由は、長期的な自己調節(autoregulation)がほぼ完全に発達し、局所的な血流制御メカニズムが圧力の違いに対してほぼ100%補償しているためです。その結果、高圧領域でも低圧領域でも、局所血流は圧力レベルに関係なく、組織のニーズにほぼ完全に一致する自己調節メカニズムによって制御されています。
妊娠中毒症(Preeclampsia, Toxemia of Pregnancy)による高血圧
妊娠中毒症(preeclampsia、妊娠中毒症とも呼ばれる)という症候群は、約5%〜10%の妊婦に発生します。妊娠中毒症の症状の一つに、高血圧があり、通常は出産後に収まります。妊娠中毒症の正確な原因は完全には理解されていませんが、胎盤の虚血(ischemia)と、その後の胎盤からの毒性因子の放出が、この障害の多くの症状、特に母体の高血圧の原因となると考えられています。虚血性胎盤から放出される物質は、腎臓の血管を含む全身の血管内皮細胞の機能不全を引き起こします。この内皮機能不全は、一酸化窒素(nitric oxide)やその他の血管拡張物質の放出を減少させ、血管収縮、糸球体から腎尿細管への体液濾過速度の低下、腎圧ナトリウム利尿(renal pressure natriuresis)の障害、および高血圧の発生を引き起こします。
妊娠中毒症における高血圧に寄与するもう一つの病的異常は、腎糸球体膜(glomerular membranes)の肥厚(おそらく自己免疫反応によるもの)であり、これも糸球体の体液濾過速度を低下させます。明らかな理由で、尿の正常な形成を引き起こすために必要な動脈圧のレベルが上昇し、長期的な動脈圧レベルもそれに応じて上昇します。これらの患者は特に過剰な塩摂取をした場合に高血圧になりやすいです。
神経性高血圧(Neurogenic Hypertension)
急性神経性高血圧は、交感神経系(sympathetic nervous system)の強い刺激によって引き起こされることがあります。たとえば、人が何らかの理由で興奮したり、不安な状態にあると、交感神経系が過剰に刺激され、全身に末梢血管収縮(peripheral vasoconstriction)が生じ、急性高血圧(acute hypertension)が起こります。
別のタイプの急性神経性高血圧は、圧受容器(baroreceptors)からの神経が切断されたり、延髄(medulla oblongata)両側の孤束路(tractus solitarius)が破壊されたときに発生します。これらは、頸動脈と大動脈の圧受容器からの神経が脳幹で接続する部位です。圧受容器からの通常の神経信号が突然停止すると、あたかも大動脈と頸動脈の動脈圧が突然低下したかのように、神経圧力制御メカニズムに影響を与えます。つまり、通常の圧受容器からの神経信号による血管運動中枢への抑制効果が失われ、血管運動中枢が突然非常に活発になり、平均動脈圧が100 mmHgから最大160 mmHgに増加します。約2日以内に圧力はほぼ正常に戻りますが、これは圧受容器の信号が欠けていることに対する血管運動中枢の反応が消えていくためであり、これを圧受容器圧制御メカニズムの中枢リセット(central resetting)と呼びます。したがって、圧受容器神経の切断によって引き起こされる神経性高血圧は主に急性のものであり、慢性のものではありません。
交感神経系は、腎交感神経(renal sympathetic nerves)の活性化を通じて、いくつかの慢性高血圧(chronic hypertension)の形式においても重要な役割を果たします。たとえば、体重増加や肥満はしばしば交感神経系の活性化を引き起こし、それが腎交感神経を刺激して腎圧ナトリウム利尿(renal pressure natriuresis)を妨げ、慢性高血圧を引き起こします。これらの異常は、後に説明するように、一次性(本態性)高血圧(primary or essential hypertension)の患者の多くにおいて主要な役割を果たしていると考えられています。
高血圧の遺伝的原因(Genetic Causes of Hypertension)珍しい
いくつかの動物種、例えば異なる系統のラットやウサギ、および少なくとも1系統の犬において、自然発生の遺伝性高血圧が観察されています。最も研究されているラットの系統であるオカモト自然発生高血圧ラット(Okamoto spontaneously hypertensive rat)において、高血圧の初期段階では交感神経系が通常のラットよりもかなり活発であるという証拠があります。このタイプの高血圧の後期段階では、腎臓のネフロンに構造的変化が観察されています。(1) 糸球体前腎動脈抵抗の増加、および (2) 糸球体膜の透過性の低下です。これらの構造的変化も高血圧の長期的な持続に寄与している可能性があります。他の高血圧ラット系統では、腎機能の障害も観察されています。
人間においては、高血圧を引き起こす可能性のあるいくつかの遺伝子変異が特定されています。これらの高血圧の形式は「単一遺伝子性高血圧(monogenic hypertension)」と呼ばれ、単一の遺伝子の変異によって引き起こされます。これらの遺伝的障害の興味深い特徴は、すべて腎機能の障害を引き起こすことです。これは、腎細動脈の抵抗の増加や腎尿細管による過剰な塩と水の再吸収によるものです。一部のケースでは、再吸収の増加は腎尿細管上皮細胞におけるナトリウムまたは塩化物の輸送を直接増加させる遺伝子変異によるものです。他のケースでは、遺伝子変異が腎尿細管による塩と水の再吸収を刺激するホルモンの合成や活性を増加させます。したがって、これまでに発見されたすべての単一遺伝子性高血圧性障害において、高血圧への最終的な共通経路は腎機能の障害であるようです。ただし、単一遺伝子性高血圧はまれであり、すべての既知の形式を合わせても人間の高血圧の1%未満を占めています。
一次性(本態性)高血圧(PRIMARY (ESSENTIAL) HYPERTENSION)(※原因不明という意味)
高血圧のある人の約90%から95%は一次性高血圧(primary hypertension)であると言われており、多くの臨床医によって「本態性高血圧(essential hypertension)」と呼ばれています。これらの用語は、高血圧が未知の原因によるものであることを意味します。これは、腎動脈狭窄(renal artery stenosis)や単一遺伝子性高血圧などの既知の原因に続発する高血圧の形式とは対照的です。
多くの患者において、過剰な体重増加と座りがちなライフスタイルが一次性高血圧の主要な原因として重要な役割を果たしているようです。高血圧のあるほとんどの患者は過体重であり、異なる集団の研究から、過剰な脂肪蓄積が一次性高血圧の発症リスクの65%から75%を占める可能性が示唆されています。臨床研究では、高血圧のあるほとんどの患者において、体重減少が血圧を下げる価値があることが明確に示されており、高血圧の治療ガイドラインでは、治療の第一歩として身体活動の増加と体重減少を推奨しています。
一次性高血圧の以下の特徴は、過剰な体重増加と肥満によって引き起こされます:
- 心拍出量(cardiac output)は、追加の脂肪組織に必要な追加の血流のために増加します。しかし、体重増加による代謝率の増加と臓器や組織の成長により、心臓、腎臓、消化管、および骨格筋における血流も増加します。高血圧が何ヶ月も何年も持続すると、総末梢血管抵抗(total peripheral vascular resistance)が増加することがあります。
- 過体重の患者では交感神経活動(sympathetic nerve activity)、特に腎臓における活動が増加しています。肥満者における交感神経活動の増加の原因は完全には理解されていませんが、脂肪細胞から放出されるホルモン(例えばレプチン(leptin))が視床下部(hypothalamus)の複数の領域を直接刺激し、それが脳の延髄(medulla)の血管運動中枢に興奮性の影響を与えることが示唆されています。また、肥満者、特に閉塞性睡眠時無呼吸(obstructive sleep apnea)を持つ人々では、動脈圧受容器(arterial baroreceptors)の感受性が低下していることや化学受容器(chemoreceptors)の活性化の証拠もあります。
- 多くの肥満患者ではアンジオテンシンIIおよびアルドステロンのレベルが増加しています。この増加は部分的に交感神経刺激の増加によって引き起こされ、これが腎臓からのレニン分泌を増加させ、その結果としてアンジオテンシンIIが形成され、それが副腎(adrenal gland)を刺激してアルドステロンを分泌させることが原因かもしれません。
- 腎圧ナトリウム利尿(renal-pressure natriuresis)メカニズムが障害されており、動脈圧が高くなるか腎機能が何らかの形で改善されない限り、腎臓は適切な量の塩と水を排泄しません。本態性高血圧の人の平均動脈圧が150 mmHgである場合、平均動脈圧を正常な100 mmHgに急激に低下させても(ただし圧力の低下以外に腎機能を変えない場合)、ほぼ完全な無尿が引き起こされます。その後、圧力が150 mmHgに戻るまで塩と水を保持します。しかし、有効な抗高血圧治療による慢性的な動脈圧の低下は、これらの治療が腎圧ナトリウム利尿を改善するため、通常、腎臓による塩と水の保持を顕著に引き起こすことはありません。
- 肥満動物および肥満患者における実験的研究は、肥満高血圧における腎圧ナトリウム利尿の障害が主に、交感神経活動の増加およびアンジオテンシンIIとアルドステロンのレベルの増加による腎尿細管での塩と水の再吸収の増加によるものであることを示唆しています。しかし、高血圧が効果的に治療されない場合、腎臓に血管損傷が生じ、糸球体濾過率(glomerular filtration rate)が低下し、高血圧の重症度が増加する可能性もあります。最終的に、肥満および関連する代謝障害に関連したコントロールされていない高血圧は、重度の血管損傷および腎機能の完全な喪失につながる可能性があります。
本態性高血圧における動脈圧制御のグラフィック分析(Graphic Analysis of Arterial Pressure Control in Essential Hypertension)
図19-15は、本態性高血圧(essential hypertension)のグラフィック分析です。この図の曲線は「ナトリウム負荷腎機能曲線(sodium-loading renal function curves)」と呼ばれています。これは、各ケースにおいて動脈圧を非常にゆっくりと、数日または数週間にわたってナトリウム摂取量を徐々に増加させることによって増加させることで得られます。ナトリウム負荷タイプの曲線は、数日ごとにナトリウム摂取量を新しいレベルに増やし、腎臓のナトリウム排出量が摂取量とバランスするのを待ち、その間に動脈圧の変化を記録することによって決定できます。
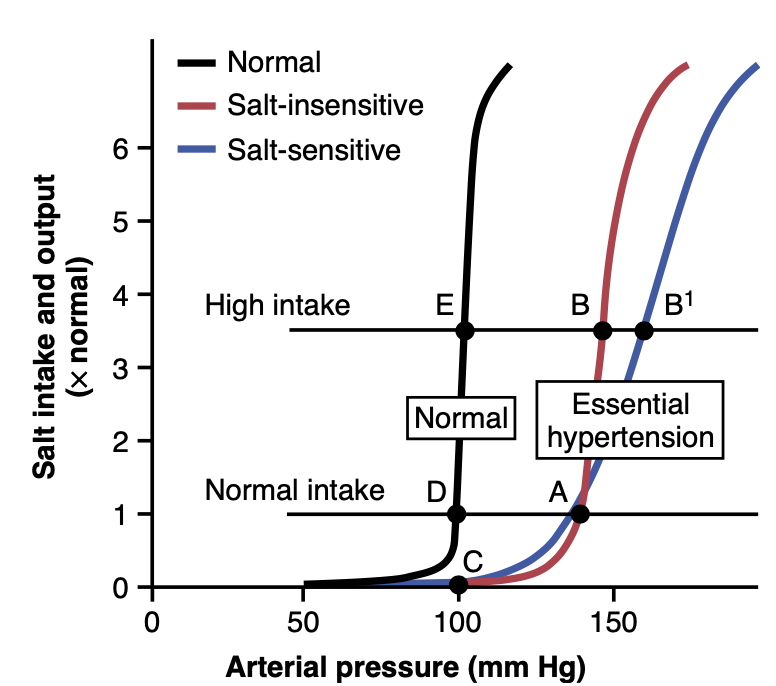
Figure 19-15. Analysis of arterial pressure regulation in (1) salt- insensitive essential hypertension and (2) salt-sensitive essential hy- pertension. (Modified from Guyton AC, Coleman TG, Young DB, et al: Salt balance and long-term blood pressure control. Annu Rev Med 31:15, 1980.)
この手順を本態性高血圧の患者に使用すると、図19-15の右側に示されている2つのタイプの曲線が記録できます。一つは「塩感受性が低い高血圧(salt-insensitive hypertension)」で、もう一つは「塩感受性が高い高血圧(salt-sensitive hypertension)」です。いずれの場合も、曲線は右にシフトし、正常な動脈圧の人に比べて高い圧力レベルに移動しています。塩感受性が低い本態性高血圧の場合、通常の塩摂取から高塩摂取に変更しても動脈圧は大幅には増加しません。しかし、塩感受性が高い本態性高血圧の患者では、高塩摂取が高血圧を顕著に悪化させます。
さらに2つの点を強調すべきです。まず、血圧の塩感受性は全か無かの所見ではなく、定量的であり、個々の感受性には違いがあります。次に、血圧の塩感受性は固定された特性ではなく、通常、年齢が増すと特に50〜60歳以降に塩感受性が増加します。これは腎臓の機能単位(ネフロン)の数が徐々に減少し始めるためです。
塩感受性が低い本態性高血圧と塩感受性が高い高血圧の違いは、これら2つの高血圧タイプの患者の腎臓の構造的または機能的な違いに関連していると考えられます。例えば、塩感受性高血圧は、ネフロンの漸進的な喪失や通常の加齢のために、さまざまな慢性腎疾患とともに発生することがあります(第32章で説明)。また、レニン–アンジオテンシン系の異常な機能も、以前に説明したように、動脈圧を塩感受性にする原因となることがあります。
本態性高血圧の治療(Treatment of Essential Hypertension)
まず、現在の高血圧治療のガイドラインでは、ほとんどの患者において身体活動の増加と体重減少を目的とした生活習慣の改善を推奨しています。残念ながら、多くの患者は体重を減らすことができず、降圧薬による薬物治療が開始される必要があります。高血圧治療に使用される薬には2つの一般的なクラスがあります:(1) 腎血流と糸球体濾過率を増加させる血管拡張薬(vasodilator drugs)、および (2) 塩と水の尿細管再吸収を減少させるナトリウム利尿薬または利尿薬(natriuretic or diuretic drugs)です。
血管拡張薬は通常、腎臓だけでなく体内の多くの組織でも血管拡張を引き起こします。異なる血管拡張薬は、以下のいずれかの方法で作用します:(1) 腎臓への交感神経信号を抑制するか、腎血管および腎尿細管に対する交感神経伝達物質の作用をブロックすることによって;(2) 腎血管の平滑筋を直接弛緩させることによって;または (3) 腎血管および腎尿細管に対するレニン–アンジオテンシン–アルドステロン系の作用をブロックすることによって。
腎尿細管による塩と水の再吸収を減少させる薬には、特に、尿細管壁を通じたナトリウムの能動輸送をブロックする薬が含まれます。このブロックにより、水の再吸収も防止されることが、第32章で詳しく説明されています。これらのナトリウム利尿薬または利尿薬については、後の章でさらに詳しく説明します。
動脈圧制御のための統合された多面的システムの概要
動脈圧が単一の圧力制御システムによって規制されているのではなく、特定の機能を果たす複数の相互に関連するシステムによって規制されていることは明らかです。例えば、人が激しく出血して圧力が突然低下した場合、圧力制御システムには2つの問題があります。1つは生存であり、動脈圧を急速に十分なレベルまで戻して、その人が急性エピソードを乗り切ることです。もう1つは、血液量と動脈圧を最終的に正常なレベルに戻し、循環システムが完全な正常性を再確立できるようにすることです。
第18章で見たように、動脈圧の急性変化に対する最初の防御線は神経制御システムです。本章では、動脈圧の長期的な制御を主に腎メカニズムによって達成する第2の防御線を強調しました。しかし、他にもパズルのピースがあります。図19-16はこれらのピースをまとめるのに役立ちます。
図19-16は、8つの動脈圧制御メカニズムの即時(秒と分)および長期(時間と日)の制御反応をフィードバックゲインとして示しています。これらのメカニズムは3つのグループに分けられます:(1) 秒または分以内に反応するもの;(2) 中間の時間期間で反応するもの、つまり分または時間;(3) 日、月、および年単位で長期的な動脈圧調整を行うもの。
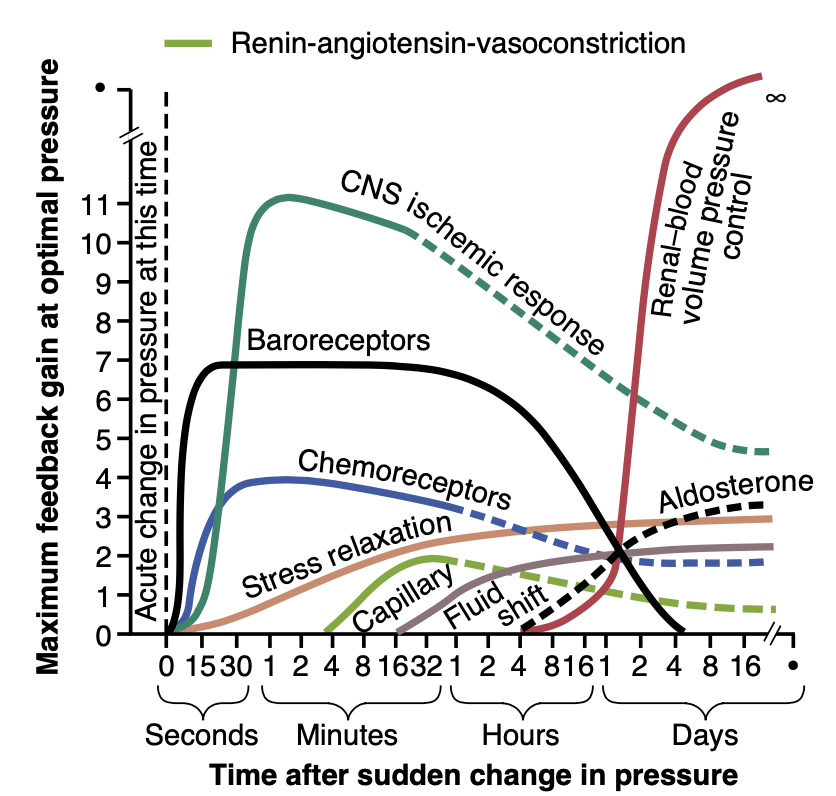
Figure 19-16. Approximate potency of various arterial pressure control mechanisms at different time intervals after the onset of a disturbance to the arterial pressure. Note especially the near-infinite gain (∞) of the renal body fluid pressure control mechanism that oc- curs after a few weeks’ time. CNS, Central nervous system. (Modified from Guyton AC: Arterial Pressure and Hypertension. Philadelphia: WB Saunders, 1980.)
数秒または数分以内に作用する動脈圧制御メカニズム
迅速に作用する圧力制御メカニズムは、ほぼすべてが急性神経反射または他の自律神経系の反応です。図19-16では、以下の3つのメカニズムが数秒以内に反応することが示されています:
(1) 圧受容器フィードバックメカニズム(baroreceptor feedback mechanism);
(2) 中枢神経系虚血メカニズム(central nervous system ischemic mechanism);および
(3) 化学受容器メカニズム(chemoreceptor mechanism)。
これらのメカニズムは数秒以内に反応を開始するだけでなく、強力でもあります。圧力が急激に低下した場合、例えば重度の出血によって引き起こされた場合、神経メカニズムは以下のように組み合わせて作用します:
(1) 静脈を収縮させ、血液を心臓に移動させる;
(2) 心拍数と心臓の収縮力を増加させ、心臓のポンプ能力を向上させる;
(3) ほとんどの末梢動脈を収縮させる。
これらすべての効果はほぼ瞬時に起こり、動脈圧を生存範囲に戻します。圧力が突然高くなりすぎた場合、例えば急速な過剰輸血に応じて同じ制御メカニズムが逆方向に作用し、再び圧力を正常に戻します。
数分後に作用する動脈圧制御メカニズム
急性の動脈圧変化の後、数分経ってから反応を示すいくつかの圧力制御メカニズムがあります。図19-16に示されている3つのメカニズムは次のとおりです:(1) レニン–アンジオテンシン血管収縮メカニズム(renin-angiotensin vasoconstrictor mechanism);(2) 血管のストレス緩和(stress relaxation)メカニズム;および (3) 組織毛細血管壁を通じた体液の移動により、必要に応じて血液量を再調整するメカニズムです。
レニン–アンジオテンシン血管収縮システムの役割については、必要に応じて動脈圧を増加させるための準急性の手段として十分に説明しました。ストレス緩和メカニズムは次の例で示されます。血管内圧が高すぎると、血管は数分から数時間にわたってますます伸展し、その結果、血管内圧は正常に向かって低下します。このような血管の持続的な伸展を「ストレス緩和(stress relaxation)」と呼び、これは中期的な圧力の「緩衝」として機能します。
毛細血管体液移動メカニズムとは、毛細血管圧が低すぎる場合、組織から体液が毛細血管膜を通じて循環に吸収され、血液量が増加し、循環の圧力が上昇することを意味します。逆に、毛細血管圧が高すぎると、体液は循環から組織に移動し、それにより血液量と循環全体の圧力が低下します。
これらの中間的なメカニズムはほとんどが30分から数時間以内に活性化されます。この期間中、神経メカニズムは通常、徐々に効果を失い、これらの非神経的な中期的圧力制御手段の重要性が示されます。
動脈圧の長期的な調整メカニズム
本章の目的は、動脈圧の長期的な制御における腎臓の役割を説明することでした。図19-16の最も右に示されているのは、腎血液量圧制御メカニズム(renal–blood volume pressure control mechanism)であり、これは腎体液圧制御メカニズム(renal–body fluid pressure control mechanism)と同じです。これは、数時間後に顕著な反応を示し始めることを示しています。しかし、最終的には動脈圧の制御に対してほぼ無限に近いフィードバックゲインを発展させます。これは、このメカニズムが最終的に動脈圧をほぼ完全に、正常な塩分と水の排出を提供する圧力レベルに戻すことができることを意味します。
腎体液メカニズムの圧力調整レベルに影響を与える要因はいくつもあります。図19-16に示されている要因の一つはアルドステロン(aldosterone)です。動脈圧の低下は数分以内にアルドステロン分泌の増加につながり、次の数時間から数日間にわたり、この効果が腎体液メカニズムの圧力制御特性を修正する重要な役割を果たします。
特に重要なのは、レニン–アンジオテンシン系(renin-angiotensin system)とアルドステロンおよび腎体液メカニズムとの相互作用です。例えば、人の塩摂取は日によって非常に大きく変動します。本章で見たように、塩摂取は通常の10分の1に減少することもあれば、10倍から15倍に増加することもありますが、レニン–アンジオテンシン–アルドステロン系が完全に機能している場合、平均動脈圧の調整レベルは数mmHgしか変化しません。しかし、機能するレニン–アンジオテンシン–アルドステロン系がない場合、血圧は塩摂取の変化に非常に敏感になります。
したがって、動脈圧の制御は、神経圧制御の生命を救う措置から始まり、中期的圧制御の持続的な特性に続き、最終的に腎体液メカニズムによって長期的な圧力レベルで安定化されます。この長期的なメカニズムは、さらにレニン–アンジオテンシン–アルドステロン系、神経系、および特定の目的のために特別な血圧制御能力を提供するいくつかの他の要因との複数の相互作用を持っています。
In addition to the rapidly acting mechanisms for regula- tion of arterial pressure discussed in Chapter 18, the body also has powerful mechanisms for regulating arterial pressure week after week and month after month. This long-term control of arterial pressure is closely inter- twined with homeostasis of body fluid volume, which is determined by the balance between fluid intake and out- put. For long-term survival, fluid intake and output must be precisely balanced, a task that is performed by multiple nervous and hormonal controls and by local control sys- tems in the kidneys that regulate their excretion of salt and water. In this chapter, we discuss these renal–body fluid systems that play a major role in long-term blood pressure regulation.
RENAL–BODY FLUID SYSTEM FOR ARTERIAL PRESSURE CONTROL
The renal–body fluid system for arterial pressure control acts slowly but powerfully, as follows. If blood volume increases and vascular capacitance is not altered, arterial pressure will also increase. The rising pressure, in turn, causes the kidneys to excrete the excess volume, thus returning the pressure back toward normal.
In the phylogenetic history of animal development, this renal–body fluid system for pressure control is a primitive one. It is fully operative in one of the lowest of vertebrates, the hagfish. This animal has a low arterial pressure, only 8 to 14 mm Hg, and this pressure increases almost directly in proportion to its blood volume. The hagfish continually drinks sea water, which is absorbed into its blood, increas- ing the blood volume and blood pressure. However, when the pressure rises too high, the kidney excretes the excess volume into the urine and relieves the pressure. At low pressure, the kidney excretes less fluid than is ingested. Therefore, because the hagfish continues to drink, extra- cellular fluid volume, blood volume, and pressure all build up again to the higher levels.
This primitive mechanism of pressure control has sur- vived throughout the ages, but with the addition of multi- ple nervous system, hormones, and local control systems that also contribute to the regulation of salt and water excretion. In humans, kidney output of water and salt is
just as sensitive—if not more so—to pressure changes as in the hagfish. Indeed, an increase in arterial pressure in the human of only a few millimeters of Hg can double the renal output of water, a phenomenon called pressure diuresis, as well as double the output of salt, called pres- sure natriuresis.
In humans, just as in the hagfish, the renal–body fluid system for arterial pressure control is a fundamen- tal mechanism for long-term arterial pressure control. However, through the stages of evolution, multiple refine- ments have been added to make this system much more precise in its control. An especially important refinement, as discussed later, has been the addition of the renin- angiotensin mechanism.
QUANTITATION OF PRESSURE DIURESIS AS A BASIS FOR ARTERIAL PRESSURE CONTROL
Figure 19-1 shows the approximate average effect of dif- ferent arterial pressure levels on the renal output of salt and water by an isolated kidney, demonstrating mark- edly increased urine output as the pressure rises. This increased urinary output is the phenomenon of pressure diuresis. The curve in this figure is called a renal urinary output curve or a renal function curve. In humans, at an arterial pressure of 50 mm Hg, the urine output is essen- tially zero. At 100 mm Hg, it is normal and, at 200 mm Hg, it is 4 to 6 times normal. Furthermore, not only does increasing the arterial pressure increase urine volume output, but it also causes an approximately equal increase in sodium output, which is the phenomenon of pressure natriuresis.
Experiment Demonstrating the Renal–Body Fluid System for Arterial Pressure Control. Figure 19-2 shows the results of an experiment in dogs in which all the nervous reflex mechanisms for blood pressure con- trol were first blocked. Then, the arterial pressure was suddenly elevated by infusing about 400 ml of blood intravenously. Note the rapid increase in cardiac out- put to about double normal and the increase in mean arterial pressure to 205 mm Hg, 115 mm Hg above its resting level. Shown by the middle curve is the effect of this increased arterial pressure on urine output, which increased 12-fold. Along with this tremendous loss of fluid in the urine, both the cardiac output and arterial pressure returned to normal during the subsequent hour. Thus, one sees an extreme capability of the kidneys to eliminate excess fluid volume from the body in response to high arterial pressure and, in so doing, to return the arterial pressure back to normal.
Renal–Body Fluid Mechanism Provides Nearly Infinite Feedback Gain for Long-term Arterial Pressure Control. Figure 19-1 shows the relationship of the following: (1) the renal output curve for water and salt in response to rising ar- terial pressure; and (2) the line that represents the net water and salt intake. Over a long period, the water and salt output must equal the intake. Furthermore, the only point on the graph in Figure 19-1 at which output equals intake is where the two curves intersect, called the equilibrium point (point A). Let us see what happens if the arterial pressure increases above or decreases below the equilibrium point.
First, assume that the arterial pressure rises to 150 mm Hg (point B). At this level, the renal output of water and salt is almost three times as great as intake. Therefore, the body loses fluid, the blood volume decreases, and the arterial pressure decreases. Furthermore, this negative balance of fluid will not cease until the pressure falls all the way back exactly to the equilibrium level. Even when the arterial pressure is only a few mm Hg greater than the equilibrium level, there still is slightly more loss of water and salt than intake, so the pressure continues to fall that last few mm Hg until the pressure eventually returns to the equilibrium point.
If the arterial pressure falls below the equilibrium point, the intake of water and salt is greater than the output. Therefore, body fluid volume increases, blood volume increases, and the arterial pressure rises until once again it returns to the equilibrium point. This return of the arterial pressure always back to the equilibrium point is known as the near-infinite feedback gain principle for control of arterial pressure by the renal–body fluid mechanism.
Two Key Determinants of Long-Term Arterial Pressure.
In Figure 19-1, one can also see that two basic long-term factors determine the long-term arterial pressure level. As long as the two curves representing the renal output of salt and water and the intake of salt and water remain exactly as they are shown in Figure 19-1, the mean arte- rial pressure level will eventually readjust to 100 mm Hg, which is the pressure level depicted by the equilibrium point of this figure. Furthermore, there are only two ways in which the pressure of this equilibrium point can be changed from the 100 mm Hg level. One is by shifting the pressure level of the renal output curve for salt and water, and the other is by changing the level of the water and salt intake line. Therefore, expressed simply, the two primary determinants of the long-term arterial pressure level are as follows:
1. The degree of pressure shift of the renal output curve for water and salt
2. The level of the water and salt intake
Operation of these two determinants in the control of arterial pressure is demonstrated in Figure 19-3. In Fig- ure 19-3A, some abnormality of the kidneys has caused the renal output curve to shift 50 mm Hg in the high- pressure direction (to the right). Note that the equilib- rium point has also shifted to 50 mm Hg higher than normal. Therefore, one can state that if the renal output curve shifts to a new pressure level, the arterial pressure will follow to this new pressure level within a few days.
Figure 19-3B shows how a change in the level of salt and water intake also can change the arterial pressure. In this case, the intake level has increased fourfold, and the equilibrium point has shifted to a pressure level of 160 mm Hg, 60 mm Hg above the normal level. Conversely, a decrease in the intake level would reduce the arterial pressure.
Thus, it is impossible to change the long-term mean arte- rial pressure level to a new value without changing one or both of the two basic determinants of long-term arterial pressure, either (1) the level of salt and water intake or (2) the degree of shift of the renal function curve along the pressure axis. However, if either of these is changed, one finds the arterial pressure thereafter to be regulated at a new pressure level, the arterial pressure at which the two new curves intersect.
In most people, however, the renal function curve is much steeper than that shown in Figure 19-3, and changes in salt intake have only a modest effect on arterial pressure, as discussed in the next section.
Chronic Renal Output Curve Much Steeper Than the Acute Curve. An important characteristic of pressure natriuresis (and pressure diuresis) is that chronic changes in arterial pressure, lasting for days or months, have a much greater effect on the renal output of salt and wa- ter than that observed during acute changes in pressure (Figure 19-4). Thus, when the kidneys are functioning normally, the chronic renal output curve is much steeper than the acute curve.
The powerful effects of chronic increases in arte- rial pressure on urine output occur because increased pressure not only has direct hemodynamic effects on the kidney to increase excretion, but also has indirect effects mediated by nervous and hormonal changes that occur when blood pressure is increased. For example, increased arterial pressure decreases activity of the sympathetic nervous system, partly through the baro- receptor reflex mechanisms discussed in Chapter 18, and by reducing formation of various hormones such as angiotensin II and aldosterone that tend to reduce salt and water excretion by the kidneys. Reduced activity of these antinatriuretic systems therefore amplifies the effectiveness of pressure natriuresis and diuresis in rais- ing salt and water excretion during chronic increases in arterial pressure (see Chapters 28 and 30 for further discussion).
Conversely, when blood pressure is reduced, the sym- pathetic nervous system is activated, and formation of antinatriuretic hormones is increased, adding to the direct effects of reduced pressure to decrease renal out- put of salt and water. This combination of direct effects of pressure on the kidneys and indirect effects of pressure on the sympathetic nervous system and various hormone systems make pressure natriuresis and diuresis extremely powerful factors for long-term control of arterial pressure and body fluid volumes.
The importance of neural and hormonal influences on pressure natriuresis is especially evident during chronic changes in sodium intake. If the kidneys and nervous and hormonal mechanisms are functioning normally, chronic increases in intakes of salt and water to as high as six times normal are usually associated with little effect on arterial pressure. Note that the blood pressure equilibrium point B on the curve is nearly the same as point A, the equilibrium point at normal salt intake. Conversely, decreases in salt and water intake to as low as one-sixth normal typically have little effect on arterial pressure. Thus, many per- sons are said to be salt-insensitive because large varia- tions in salt intake do not change blood pressure more than a few mm Hg.
Persons with kidney injury or excessive secretion of antinatriuretic hormones such as angiotensin II or aldo- sterone, however, may be salt-sensitive, with an attenu- ated renal output curve similar to the acute curve shown in Figure 19-4. In these cases, even moderate increases in salt intake may cause significant increases in arterial pressure.
Some of the factors that cause blood pressure to be salt-sensitive include loss of functional nephrons due to kidney injury and excessive formation of antinatri- uretic hormones such as angiotensin II or aldosterone. For example, surgical reduction of kidney mass or injury to the kidney due to hypertension, diabetes, or various kidney diseases all cause blood pressure to be more sensitive to changes in salt intake. In these cases, greater than normal increases in arterial pressure are required to raise renal output sufficiently to maintain a balance between the intake and output of salt and water.
There is evidence that long-term high salt intake, last- ing for several years, may actually damage the kidneys and eventually makes blood pressure more salt-sensitive. We will discuss salt sensitivity of blood pressure in patients with hypertension later in this chapter.
Failure of Increased Total Peripheral Resistance to Elevate Long-Term Level of Arterial Pressure if Fluid Intake and Renal Function Do Not Change
Recalling the basic equation for arterial pressure—arte- rial pressure equals cardiac output times total peripheral resistance—it is clear that an increase in total peripheral resistance should elevate the arterial pressure. Indeed, when the total peripheral resistance is acutely increased, the arterial pressure does rise immediately. Yet, if the kidneys continue to function normally, the acute rise in arterial pressure usually is not maintained. Instead, the arterial pressure returns all the way to normal within about 1 or 2 days. Why?
The reason for this phenomenon is that increasing vas- cular resistance everywhere else in the body besides in the kidneys does not change the equilibrium point for blood pressure control as dictated by the kidneys (see Figures 19-1 and 19-3). Instead, the kidneys immediately begin to respond to the high arterial pressure, causing pres- sure diuresis and pressure natriuresis. Within hours, large amounts of salt and water are lost from the body; this pro- cess continues until the arterial pressure returns to the equilibrium pressure level. At this point, blood pressure is normalized, and extracellular fluid volume and blood volume are decreased to levels below normal.
Figure 19-5 shows the approximate cardiac outputs and arterial pressures in different clinical conditions in which the long-term total peripheral resistance is much less than or much greater than normal, but kidney excre- tion of salt and water is normal. Note in all these differ- ent clinical conditions that the arterial pressure is also normal.
A word of caution is necessary at this point in our discussion. Often, when the total peripheral resistance increases, this also increases the intrarenal vascular resistance at the same time, which alters the function of the kidney and can cause hypertension by shifting the renal function curve to a higher pressure level, as shown in Figure 19-3A. We will see an example of this mecha- nism later in this chapter when we discuss hypertension caused by vasoconstrictor mechanisms. However, it is the increase in renal resistance that is the culprit, not the increased total peripheral resistance—an important distinction.
Increased Fluid Volume Can Elevate Arterial Pressure by Increasing Cardiac Output or Total Peripheral Resistance
The overall mechanism whereby increased extracellular fluid volume may elevate arterial pressure, if vascular capacity is not simultaneously increased, is shown in Figure 19-6. The sequential events are as follows: (1) increased extracellular fluid volume, which (2) increases the blood volume, which (3) increases the mean circulatory filling pressure, which (4) increases venous return of blood to the heart, which (5) increases cardiac output, which (6) increases arterial pres- sure. The increased arterial pressure, in turn, increases the renal excretion of salt and water and may return extracellular fluid volume to nearly normal if kidney function is normal and vascular capacity is unaltered.
Note especially in this case the two ways in which an increase in cardiac output can increase the arterial pres- sure. One of these is the direct effect of increased car- diac output to increase the pressure, and the other is an indirect effect to raise total peripheral vascular resistance through autoregulation of blood flow. The second effect can be explained as follows.
Referring to Chapter 17, let us recall that whenever an excess amount of blood flows through a tissue, the local tissue vasculature constricts and decreases the blood flow back toward normal. This phenomenon is called autoreg- ulation, which simply means regulation of blood flow by the tissue itself. When increased blood volume raises the cardiac output, blood flow tends to increase in all tissues of the body; if the increased blood flow exceeds the meta- bolic needs of the tissues, the autoregulation mechanisms constricts blood vessels all over the body, which in turn increases the total peripheral resistance.
Finally, because arterial pressure is equal to cardiac output times total peripheral resistance, the secondary increase in total peripheral resistance that results from the autoregulation mechanism helps increase the arte- rial pressure. For example, only a 5% to 10% increase in cardiac output can increase the arterial pressure from the normal mean arterial pressure of 100 mm Hg up to 150 mm Hg when accompanied by an increase in total peripheral resistance due to tissue blood flow autoregu- lation or other factors that cause vasoconstriction. The slight increase in cardiac output is often not measurable.
Importance of Salt (NaCl) in the Renal– Body Fluid Schema for Arterial Pressure Regulation
Although the discussions thus far have emphasized the importance of volume in regulation of arterial pressure, experimental studies have shown that an increase in salt intake is far more likely to elevate the arterial pres- sure, especially in people who are salt-sensitive, than is an increase in water intake. The reason for this finding is that pure water is normally excreted by the kidneys almost as rapidly as it is ingested, but salt is not excreted so easily. As salt accumulates in the body, it also indi- rectly increases the extracellular fluid volume for two basic reasons:
1. Although some additional sodium may be stored in the tissues when salt accumulates in the body, ex- cess salt in the extracellular fluid increases the fluid osmolality. The increased osmolarity stimulates the thirst center in the brain, making the person drink extra amounts of water to return the extracellular salt concentration to normal and increasing the ex- tracellular fluid volume.
2. The increase in osmolality caused by the excess salt in the extracellular fluid also stimulates the hypo- thalamic–posterior pituitary gland secretory mech- anism to secrete increased quantities of antidiuretic hormone (discussed in Chapter 29). The antidiu- retic hormone then causes the kidneys to reabsorb greatly increased quantities of water from the renal tubular fluid, thereby diminishing the excreted vol- ume of urine but increasing the extracellular fluid volume.
Thus, the amount of salt that accumulates in the body is an important determinant of the extracellular fluid volume. Relatively small increases in extracellular fluid and blood volume can often increase the arterial pressure substantially. This is true, however, only if the excess salt accumulation leads to an increase in blood volume and if vascular capacity is not simultaneously increased. As discussed previously, increasing salt intake in the absence of impaired kidney function or excessive formation of antinatriuretic hormones usually does not increase arterial pressure much because the kidneys rap- idly eliminate the excess salt, and blood volume is hardly altered.
CHRONIC HYPERTENSION (HIGH BLOOD PRESSURE) CAUSED BY IMPAIRED RENAL FUNCTION
When a person is said to have chronic hypertension (or high blood pressure), this means that his or her mean arterial pressure is greater than the upper range of the accepted normal measure. A mean arterial pressure greater than 110 mm Hg (normal is ≈90 mm Hg) is con- sidered to be hypertensive. (This level of mean pressure occurs when the diastolic blood pressure is greater than ≈90 mm Hg and the systolic pressure is greater than ≈135 mm Hg.) In persons with severe hypertension, the mean arterial pressure can rise to 150 to 170 mm Hg, with dia- stolic pressure as high as 130 mm Hg and systolic pressure occasionally as high as 250 mm Hg.
Even moderate elevation of arterial pressure leads to shortened life expectancy. At severely high pressures— that is, mean arterial pressures 50% or more above nor- mal—a person can expect to live no more than a few more years unless appropriately treated. The lethal effects of hypertension are caused mainly in three ways:
1. Excess workload on the heart leads to early heart failure and coronary heart disease, often causing death as a result of a heart attack.
- The high pressure frequently damages a major blood vessel in the brain, followed by death of major portions of the brain; this occurrence is a cerebral infarct. Clinically, it is called a stroke. Depending on which part of the brain is involved, a stroke can be fatal or cause paralysis, dementia, blindness, or multiple other serious brain disorders.
- High pressure almost always causes injury in the kidneys, producing many areas of renal destruction and, eventually, kidney failure, uremia, and death.
Lessons learned from the type of hypertension called volume-loading hypertension have been crucial in
understanding the role of the renal–body fluid volume mechanism for arterial pressure regulation. Volume- loading hypertension means hypertension caused by excess accumulation of extracellular fluid in the body, some examples of which follow.
Experimental Volume-Loading Hypertension Caused by Reduced Kidney Mass and Increased Salt Intake. Figure 19-7 shows a typical experiment dem- onstrating volume-loading hypertension in a group of dogs with 70% of their kidney mass removed. At the first circled point on the curve, the two poles of one of the kidneys were removed, and at the second circled point, the entire opposite kidney was removed, leaving the an- imals with only 30% of their normal renal mass. Note that removal of this amount of kidney mass increased the arterial pressure by an average of only 6 mm Hg. Then, the dogs were given salt solution to drink instead of water. Because salt solution fails to quench the thirst, the dogs drank two to four times the normal amounts of volume, and within a few days, their average arterial pressure rose to about 40 mm Hg above normal. After 2 weeks, the dogs were given tap water again instead of salt solution; the pressure returned to normal within 2 days. Finally, at the end of the experiment, the dogs were given salt solution again, and this time the pressure rose much more rapidly to a high level, again demonstrating volume-loading hypertension.
If one considers again the basic determinants of long- term arterial pressure regulation, it is apparent why hypertension occurred in the volume-loading experiment illustrated in Figure 19-7. First, reduction of the kidney mass to 30% of normal greatly reduced the ability of the kidneys to excrete salt and water. Therefore, salt and water accumulated in the body and, in a few days, raised the arterial pressure high enough to excrete the excess salt and water intake.
Sequential Changes in Circulatory Function During Development of Volume-Loading Hypertension. It is especially instructive to study the sequential changes in circulatory function during progressive development of volume-loading hypertension (Figure 19-8). A week or so before the point labeled “0” days, the kidney mass had already been decreased to only 30% of normal. Then, at this point, the intake of salt and water was increased to about six times normal and kept at this high intake there- after. The acute effect was to increase extracellular fluid volume, blood volume, and cardiac output to 20% to 40% above normal. Simultaneously, the arterial pressure began to rise but not nearly so much at first as the fluid volumes and cardiac output. The reason for this slower rise in pres- sure can be discerned by studying the total peripheral resistance curve, which shows an initial decrease in total peripheral resistance. This decrease was caused by the baroreceptor mechanism discussed in Chapter 18, which transiently attenuated the rise in pressure. However, after 2 to 4 days, the baroreceptors adapted (reset) and were no longer able to prevent the rise in pressure. At this time, the arterial pressure had risen almost to its full height because of the increase in cardiac output, even though the total peripheral resistance was still almost at the normal level.
After these early acute changes in the circulatory vari- ables had occurred, more prolonged secondary changes occurred during the next few weeks. Especially important was a progressive increase in total peripheral resistance, while at the same time the cardiac output decreased back toward normal, at least partly as a result of the long-term blood flow autoregulation mechanism discussed in Chap- ter 17 and earlier in this chapter. That is, after the car- diac output had risen to a high level and had initiated the hypertension, the excess blood flow through the tissues then caused progressive constriction of the local arteri- oles, thus returning the local blood flow in the body tis- sues and also the cardiac output toward normal while simultaneously causing a secondary increase in total peripheral resistance.
Note that the extracellular fluid volume and blood vol- ume also returned toward normal along with the decrease in cardiac output. This outcome resulted from two fac- tors. First, the increase in arteriolar resistance decreased the capillary pressure, which allowed the fluid in the tis- sue spaces to be absorbed back into the blood. Second, the elevated arterial pressure now caused the kidneys to excrete the excess volume of fluid that had initially accu- mulated in the body.
Several weeks after the initial onset of volume loading, the following effects were found:
1. Hypertension
2. Marked increase in total peripheral resistance
3. Almost complete return of the extracellular fluid
volume, blood volume, and cardiac output back to
normal
Therefore, we can divide volume-loading hypertension
into two sequential stages. The first stage results from increased fluid volume causing increased cardiac output. This increase in cardiac output mediates the hyperten- sion. The second stage in volume-loading hypertension is characterized by high blood pressure and high total peripheral resistance but return of the cardiac output so close to normal that the usual measuring techniques frequently cannot detect an abnormally elevated cardiac output.
Thus, the increased total peripheral resistance in volume-loading hypertension occurs after the hyper- tension has developed and, therefore, is secondary to the hypertension rather than being the cause of the hypertension.
Volume-Loading Hypertension in Patients Who Have No Kidneys but Are Being Maintained With an Artificial Kidney
When a patient is maintained with an artificial kidney, it is especially important to keep the patient’s body fluid volume at a normal level by removing the appropriate amount of water and salt each time the patient undergoes dialysis. If this step is not performed, and extracellular fluid volume is allowed to increase, hypertension almost invariably develops in exactly the same way as shown in Figure 19-8. That is, the cardiac output increases at first and causes hypertension. Then, the autoregulation mechanism returns the cardiac output back toward nor- mal while causing a secondary increase in total peripheral resistance. Therefore, in the end, the hypertension appears to be a high peripheral resistance type of hypertension, although the initial cause is excess volume accumulation.
Hypertension Caused by Excess Aldosterone
Another type of volume-loading hypertension is caused by excess aldosterone in the body or, occasionally, by excesses of other types of steroids. A small tumor in one of the adrenal glands occasionally secretes large quanti- ties of aldosterone, which is the condition called primary aldosteronism. As discussed in Chapters 28 and 30, aldo- sterone increases the rate of salt and water reabsorption by the tubules of the kidneys, thereby increasing blood volume, extracellular fluid volume, and arterial pressure. If salt intake is increased at the same time, the hyperten- sion becomes even greater. Furthermore, if the condition persists for months or years, the excess arterial pressure often causes pathological changes in the kidneys that make the kidneys retain even more salt and water in addi- tion to that caused directly by the aldosterone. Therefore, the hypertension often finally becomes severe to the point of being lethal.
Here again, in the early stages of this type of hyperten- sion, cardiac output is often increased but, in later stages, the cardiac output generally returns almost to normal while total peripheral resistance becomes secondarily elevated, as explained earlier in the chapter for primary volume-loading hypertension.
ROLE OF THE RENIN-ANGIOTENSIN SYSTEM IN ARTERIAL PRESSURE CONTROL
Aside from the capability of the kidneys to control arterial pressure through changes in extracellular fluid volume, the kidneys also have another powerful mechanism for controlling pressure, the renin-angiotensin system.
Renin is a protein enzyme released by the kidneys when the arterial pressure falls too low. In turn, it raises the arterial pressure in several ways, thus helping correct the initial fall in pressure.
COMPONENTS OF THE RENIN- ANGIOTENSIN SYSTEM
Figure 19-9 shows the main functional steps whereby the renin-angiotensin system helps regulate arterial pressure. Renin is synthesized and stored in the juxtaglomerular cells (JG cells) of the kidneys. The JG cells are modified smooth muscle cells located mainly in the walls of the afferent arterioles immediately proximal to the glomeruli. Multiple factors control renin secretion, including the sympathetic nervous system, various hormones, and local autacoids such as prostaglandins, nitric oxide, and endo- thelin. When the arterial pressure falls, the JG cells release renin by at least three major mechanisms:
1. Pressure-sensitive baroreceptors in the JG cells re- spond to decreased arterial pressure with increased release of renin.
2. Decreased sodium chloride delivery to the macula densa cells in the early distal tubule stimulates renin release (discussed further in Chapter 27)
3. Increased sympathetic nervous system activity stim- ulates renin release by activating beta-adrenergic receptors in the JG cells. Sympathetic stimulation also activates alpha-adrenergic receptors, which can increase renal sodium chloride reabsorption and reduce the glomerular filtration rate in cases of strong sympathetic activation. Increased renal sympathetic activity also enhances the sensitivity of renal baroreceptor and macula densa mechanisms for renin release.
Most of the renin enters the renal blood and then passes out of the kidneys to circulate throughout the entire body. However, small amounts of the renin do remain in the local fluids of the kidney and initiate several intrarenal functions.
Renin itself is an enzyme, not a vasoactive substance. As shown in Figure 19-9, renin acts enzymatically on another plasma protein, a globulin called renin substrate (or angiotensinogen), to release a 10–amino acid peptide, angiotensin I. Angiotensin I has mild vasoconstrictor properties but not enough to cause significant changes in circulatory function. The renin persists in the blood for 30 to 60 minutes and continues to cause formation of still more angiotensin I during this entire time.
Within a few seconds to minutes after the formation of angiotensin I, two additional amino acids are split from the angiotensin I to form the 8-amino acid peptide angiotensin II. This conversion occurs to a great extent in the lungs while the blood flows through the small vessels of the lungs, catalyzed by an enzyme called angiotensin-converting enzyme (ACE) that is present in the endothelium of the lung vessels. Other tissues such as the kidneys and blood vessels also contain ACE and therefore form angiotensin II locally.
Angiotensin Il is an extremely powerful vasocon-strictor, and it affects circulatory function in other ways as well. However, it persists in the blood only for 1 or 2 minutes because it is rapidly inactivated by multiple blood and tissue enzymes collectively called angiotensinases.
Angiotensin II has two principal effects that can elevate arterial pressure. The first of these, vasoconstriction in many areas of the body, occurs rapidly. Vasoconstriction occurs intensely in the arterioles and less so in the veins. Constriction of the arterioles increases the total peripheral resistance, thereby raising the arterial pressure, as demonstrated at the bottom of Figure 19-9. Also, the mild constriction of the veins promotes increased venous return of blood to the heart, thereby helping the heart pump against the increasing pressure.
The second principal means whereby angiotensin II increases the arterial pressure is decreased excretion of salt and water by the kidneys due to stimulation of aldosterone secretion, as well as direct effects on the kidneys. The salt and water retention by the kidneys slowly increases the extracellular fluid volume, which then increases the arterial pressure during subsequent hours and days. This long-term effect, through the direct and indirect actions of angiotensin II on the kidneys, is even more powerful than the acute vasoconstrictor mechanism in eventually raising the arterial pressure.
Rapidity and Intensity of the
Vasoconstrictor Pressure Response to the Renin-Angiotensin System
Figure 19-10 shows an experiment demonstrating the effect of hemorrhage on the arterial pressure under two separate conditions: (1) with the renin-angiotensin system functioning; and (2) after blocking the system with a renin-blocking antibody. Note that after hemorrhage-enough to cause acute decrease of the arterial pressure to 50 mm Hg–the arterial pressure rose back to 83 mm Hg when the renin-angiotensin system was functional.
Conversely, it rose to only 60 mm Hg when the renin-angiotensin system was blocked. This phenomenon shows that the renin-angiotensin system is powerful enough to return the arterial pressure at least halfway back to normal within a few minutes after severe hemorrhage. Therefore, this system can be of lifesaving service to the body, especially in circulatory shock.
Note also that the renin-angiotensin vasoconstrictor system requires about 20 minutes to become fully active. Therefore, it is somewhat slower for blood pressure control than the nervous reflexes and sympathetic norepinephrine-epinephrine system.
Angiotensin II Causes Renal Retention of Salt and Water-_-An Important Means for Long-Term Control of Arterial Pressure
Angiotensin II causes the kidneys to retain both salt and water in two major ways:
- Angiotensin I acts directly on the kidneys to cause salt and water retention.
- Angiotensin II stimulates the adrenal glands to secrete aldosterone, and the aldosterone in turn increases salt and water reabsorption by the kidney tubules.
Thus, whenever excess amounts of angiotensin I circulate in the blood, the entire long-term renal-body fluid mechanism for arterial pressure control automatically becomes set to a higher arterial pressure level than normal.
Mechanisms of the Direct Renal Effects of Angiotensin Il to Cause Renal Retention of Salt and Water. Angiotensin has several direct renal effects that make the kidneys retain salt and water. One major effect is to constrict the renal arterioles, especially the glomerular efferent ar-terioles, thereby diminishing blood flow through the kid-neys. The slow flow of blood reduces the pressure in the peritubular capillaries, which increases reabsorption of fluid from the tubules. Angiotensin II also has important direct actions on the tubular cells to increase tubular re-absorption of sodium and water, as discussed in Chapter
28. The combined effects of angiotensin II can sometimes decrease urine output to less than one-fifth normal.
Angiotensin II Increases Kidney Salt and Water Retention by Stimulating Aldosterone. Angiotensin
II is also one of the most powerful stimulators of aldosterone secretion by the adrenal glands, as we shall discuss in relation to body fluid regulation in Chapter 30 and in relation to adrenal gland function in Chapter 78. Therefore, when the renin-angiotensin system becomes activated, the rate of aldosterone secretion usually also increases; an important subsequent function of aldoster- one is to cause marked increase in sodium reabsorption by the kidney tubules, thus increasing the total body ex- tracellular fluid sodium and, as already explained, extra- cellular fluid volume. Thus, both the direct effect of an- giotensin II on the kidneys and its effect acting through aldosterone are important in long-term arterial pressure control.
Quantitative Analysis of Arterial Pressure Changes Caused by Angiotensin II. Figure 19-11 shows a quanti- tative analysis of the effect of angiotensin in arterial pres- sure control. This figure shows two renal function curves, as well as a line depicting a normal level of sodium intake. The left-hand renal function curve is that measured in dogs whose renin-angiotensin system had been blocked by an ACE inhibitor drug that blocks the conversion of angioten- sin I to angiotensin II. The right-hand curve was measured in dogs infused continuously with angiotensin II at a level about 2.5 times the normal rate of angiotensin formation in the blood. Note the shift of the renal output curve toward higher pressure levels under the influence of angiotensin II. This shift is caused by the direct effects of angiotensin II on the kidney and the indirect effect acting through aldoster- one secretion, as explained earlier.
Finally, note the two equilibrium points, one for zero angiotensin showing an arterial pressure level of 75 mm Hg, and one for elevated angiotensin showing a pressure level of 115 mm Hg. Therefore, the effect of angiotensin to cause renal retention of salt and water can have a powerful effect in promoting chronic elevation of the arterial pres- sure.
Role of the Renin-Angiotensin System in Maintaining a Normal Arterial Pressure Despite Large Variations in Salt Intake
One of the most important functions of the renin- angiotensin system is to allow a person to ingest very small or very large amounts of salt without causing major changes in extracellular fluid volume or arterial pressure. This function is explained by Figure 19-12, which shows that the initial effect of increased salt intake is to elevate the extracellular fluid volume, which tends to elevate the arterial pressure. Multiple effects of increased salt intake, including small increases in arterial pressure and pressure- independent effects, reduce the rate of renin secretion and angiotensin II formation, which then helps eliminate the additional salt with minimal increases in extracellular fluid volume or arterial pressure. Thus, the renin-angiotensin system is an automatic feedback mechanism that helps maintain the arterial pressure at or near the normal level, even when salt intake is increased. When salt intake is decreased below normal, exactly opposite effects take place.
To emphasize the efficacy of the renin-angiotensin system in controlling arterial pressure, when the system functions normally, the pressure usually rises no more than 4 to 6 mm Hg in response to as much as a 100-fold increase in salt intake (Figure 19-13). Conversely, when the usual suppression of angiotensin formation is pre- vented due to continuous infusion of small amounts of angiotensin II so that blood levels cannot decrease, the same increase in salt intake may cause the pressure to rise by 40 mm Hg or more (see Figure 19-13). When salt intake is reduced to as low as one-tenth normal, arterial pressure barely changes as long as the renin-angiotensin system functions normally. However, when angiotensin II formation is blocked with an ACE inhibitor, blood pres- sure decreases markedly as salt intake is reduced (see Figure 19-13). Thus, the renin-angiotensin system is per- haps the body’s most powerful system for accommodat- ing wide variations in salt intake with minimal changes in arterial pressure.
HYPERTENSION CAUSED BY RENIN- SECRETING TUMOR OR RENAL ISCHEMIA
Occasionally, a tumor of the renin-secreting JG cells occurs and secretes large quantities of renin, caus- ing formation of large amounts of angiotensin II. In all patients in whom this phenomenon has occurred, severe hypertension has developed. Also, when large amounts of angiotensin II are infused continuously for days or weeks into animals, similar severe long-term hyperten- sion develops.
We have already noted that angiotensin II can increase the arterial pressure in two ways:
1. By constricting the arterioles throughout the entire body, thereby increasing the total peripheral resist- ance and arterial pressure; this effect occurs within seconds after one begins to infuse large amounts of angiotensin II.
2. By causing the kidneys to retain salt and water; over a period of days, even moderate amounts of angio- tensin II can cause causes hypertension through its renal actions, the principal cause of the long-term elevation of arterial pressure.
One-Kidney Goldblatt Hypertension. When one kid- ney is removed, and a constrictor is placed on the renal artery of the remaining kidney, as shown in Figure 19- 14, the immediate effect is greatly reduced pressure in the renal artery beyond the constrictor, as demonstrated by the dashed curve in the figure Then, within seconds or minutes, the systemic arterial pressure begins to rise and continues to rise for several days. The pressure usually rises rapidly for the first hour or so, and this effect is fol- lowed by a slower additional rise during the next several days. When the systemic arterial pressure reaches its new stable pressure level, the renal arterial pressure distal to the constriction (the dashed curve in the figure) will have returned almost all the way back to normal. The hyper- tension produced in this way is called one-kidney Gold- blatt hypertension in honor of Harry Goldblatt, who first studied the important quantitative features of hyperten- sion caused by renal artery constriction.
The early rise in arterial pressure in Goldblatt hyper- tension is caused by the renin-angiotensin vasoconstrictor mechanism. That is, because of poor blood flow through the kidney after acute constriction of the renal artery, large quantities of renin are secreted by the kidney, as demonstrated by the lowermost curve in Figure 19-14, and this action increases angiotensin II and aldosterone levels in the blood. The angiotensin II, in turn, raises the arterial pressure acutely. The secretion of renin rises to a peak in about 1 or 2 hours but returns nearly to normal in 5 to 7 days because the renal arterial pressure by that time has also risen back to normal, so the kidney is no longer ischemic.
The second rise in arterial pressure is caused by reten- tion of salt and water by the constricted kidney, which is also stimulated by angiotensin II and aldosterone. In 5 to 7 days, the body fluid volume increases enough to raise the arterial pressure to its new sustained level. The quantita- tive value of this sustained pressure level is determined by the degree of constriction of the renal artery. That is, the aortic pressure must rise enough so that renal arterial pressure distal to the constrictor is high enough to cause normal urine output.
A similar scenario occurs in patients with stenosis of the renal artery of a single remaining kidney, as some- times occurs after a person receives a kidney transplant. Also, functional or pathological increases in resistance of the renal arterioles, due to atherosclerosis or exces- sive levels of vasoconstrictors, can cause hypertension through the same mechanisms as constriction of the main renal artery.
Two-Kidney Goldblatt Hypertension. Hypertension also can result when the artery to only one kidney is con- stricted while the artery to the other kidney is normal. The constricted kidney secretes renin and also retains salt and water because of decreased renal arterial pressure in this kidney. Then, the “normal” opposite kidney retains salt and water because of the renin produced by the is- chemic kidney. This renin causes increased formation of angiotensin II and aldosterone, both of which circulate to the opposite kidney and cause it also to retain salt and water. Thus, both kidneys—but for different reasons—be- come salt and water retainers. Consequently, hyperten- sion develops.
The clinical counterpart of two-kidney Goldblatt hypertension occurs when there is stenosis of a single renal artery—for example, caused by atherosclerosis—in a person who has two kidneys.
Hypertension Caused by Diseased Kidneys That Secrete Renin Chronically. Often, patchy areas of one or both kidneys are diseased and become ischemic because of local vascular constrictions or infarctions, whereas other areas of the kidneys are normal. When this situa- tion occurs, almost identical effects occur as in the two- kidney type of Goldblatt hypertension. That is, the patchy ischemic kidney tissue secretes renin, which, in turn—by acting through the formation of angiotensin II—causes the remaining kidney mass also to retain salt and water. One of the most common causes of renal hypertension, especially in older persons, is this patchy ischemic kidney disease.
Other Types of Hypertension Caused by Combinations of Volume Loading and Vasoconstriction
Hypertension in the Upper Part of the Body Caused by Coarctation of the Aorta. One out of every few thousand babies is born with pathological constriction or blockage of the aorta at a point beyond the aortic arterial branches to the head and arms but proximal to the renal arteries, a condition called coarctation of the aorta. When this oc- curs, blood flow to the lower body is carried by multiple small collateral arteries in the body wall, with much vascu- lar resistance between the upper aorta and lower aorta. As a consequence, the arterial pressure in the upper part of the body may be 40% to 50% higher than that in the lower body.
The mechanism of this upper body hypertension is al- most identical to that of one-kidney Goldblatt hyperten- sion. That is, when a constrictor is placed on the aorta above the renal arteries, the blood pressure in both kidneys falls at first, renin is secreted, angiotensin and aldosterone are formed, and hypertension occurs in the upper body. The arterial pressure in the lower body at the level of the kidneys rises approximately to normal, but high pressure persists in the upper body. The kidneys are no longer is- chemic, and thus secretion of renin and formation of angi- otensin and aldosterone return to nearly normal. Likewise, in coarctation of the aorta, the arterial pressure in the lower body is usually almost normal, whereas the pressure in the upper body is far higher than normal.
Role of Autoregulation in Hypertension Caused by Aortic Coarctation. A significant feature of hypertension caused by aortic coarctation is that blood flow in the arms, where the pressure may be 40% to 60% above normal, is almost exactly normal. Also, blood flow in the legs, where the pressure is not elevated, is almost exactly normal. How could this be, with the pressure in the upper body 40% to 60% greater than in the lower body? There are no differ- ences in vasoconstrictor substances in the blood of the up- per and lower body because the same blood flows to both areas. Likewise, the nervous system innervates both areas of the circulation similarly, so there is no reason to believe that there is a difference in nervous control of the blood vessels. The main reason is that long-term autoregulation develops so nearly completely that the local blood flow con- trol mechanisms have compensated almost 100% for the differences in pressure. The result is that in both the high- pressure area and low-pressure area, the local blood flow is controlled by local autoregulatory mechanisms almost exactly in accord with the needs of the tissue and not in accord with the level of the pressure.
Hypertension in Preeclampsia (Toxemia of Pregnancy).
A syndrome called preeclampsia (also called toxemia of pregnancy) develops in approximately 5% to 10% of expect- ant mothers. One of the manifestations of preeclampsia is hypertension that usually subsides after delivery of the baby. Although the precise causes of preeclampsia are not completely understood, ischemia of the placenta and sub- sequent release by the placenta of toxic factors are believed to play a role in causing many of the manifestations of this disorder, including hypertension in the mother. Substances released by the ischemic placenta, in turn, cause dysfunc- tion of vascular endothelial cells throughout the body, in- cluding the blood vessels of the kidneys. This endothelial dysfunction decreases release of nitric oxide and other vaso- dilator substances, causing vasoconstriction, decreased rate of fluid filtration from the glomeruli into the renal tu- bules, impaired renal pressure natriuresis, and the develop- ment of hypertension.
Another pathological abnormality that may contribute to hypertension in preeclampsia is thickening of the kid- ney glomerular membranes (perhaps caused by an auto- immune process), which also reduces the glomerular fluid filtration rate. For obvious reasons, the arterial pressure level required to cause normal formation of urine becomes elevated, and the long-term level of arterial pressure be- comes correspondingly elevated. These patients are espe- cially prone to extra degrees of hypertension when they have excess salt intake.
Neurogenic Hypertension. Acute neurogenic hyperten- sion can be caused by strong stimulation of the sympathetic nervous system. For example, when a person becomes ex- cited for any reason or during states of anxiety, the sym- pathetic system becomes excessively stimulated, peripheral vasoconstriction occurs everywhere in the body, and acute hypertension ensues.
Another type of acute neurogenic hypertension occurs when the nerves leading from the baroreceptors are cut or when the tractus solitarius is destroyed in each side of the medulla oblongata. These are the areas where the nerves from the carotid and aortic baroreceptors connect in the brain stem. The sudden cessation of normal nerve signals from the baroreceptors has the same effect on the nervous pressure control mechanisms as a sudden reduction of the arterial pressure in the aorta and carotid arteries. That is, loss of the normal inhibitory effect on the vasomotor center caused by normal baroreceptor nervous signals allows the vasomotor center suddenly to become extremely active and the mean arterial pressure to increase from 100 mm Hg to as high as 160 mm Hg. The pressure returns to nearly nor- mal within about 2 days because the response of the vaso- motor center to the absent baroreceptor signal fades away, which is called central resetting of the baroreceptor pressure control mechanism. Therefore, the neurogenic hyperten- sion caused by sectioning the baroreceptor nerves is mainly an acute type of hypertension, not a chronic type.
The sympathetic nervous system also plays an important role in some forms of chronic hypertension, in large part by activation of the renal sympathetic nerves. For example, ex- cess weight gain and obesity often lead to activation of the sympathetic nervous system, which in turn stimulates the renal sympathetic nerves, impairs renal pressure natriure- sis, and causes chronic hypertension. These abnormalities appear to play a major role in a large percentage of patients with primary (essential) hypertension, as discussed later.
Genetic Causes of Hypertension. Spontaneous heredi- tary hypertension has been observed in several strains of animals, including different strains of rats and rabbits and at least one strain of dogs. In the strain of rats that has been studied to the greatest extent, the Okamoto spontaneously hypertensive rat, there is evidence that in early develop-
ment of the hypertension, the sympathetic nervous system is considerably more active than in normal rats. In the later stages of this type of hypertension, structural changes have been observed in the nephrons of the kidneys: (1) increased preglomerular renal arterial resistance; and (2) decreased permeability of the glomerular membranes. These struc- tural changes could also contribute to the long-term con- tinuance of the hypertension. In other strains of hyperten- sive rats, impaired renal function also has been observed.
In humans, several different gene mutations have been identified that can cause hypertension. These forms of hy- pertension are called monogenic hypertension because they are caused by mutation of a single gene. An interesting fea- ture of these genetic disorders is that they all cause impaired kidney function, either by increased resistance of the renal arterioles or by excessive salt and water reabsorption by the renal tubules. In some cases, the increased reabsorption is due to gene mutations that directly increase the transport of sodium or chloride in the renal tubular epithelial cells. In other cases, the gene mutations cause increased synthesis or activity of hormones that stimulate renal tubular salt and water reabsorption. Thus, in all monogenic hypertensive disorders discovered thus far, the final common pathway to hypertension appears to be impaired kidney function. Mo- nogenic hypertension, however, is rare, and all the known forms together account for less than 1% of cases of human hypertension.
PRIMARY (ESSENTIAL) HYPERTENSION
About 90% to 95% of all people who have hypertension are said to have primary hypertension, also referred to as essential hypertension by many clinicians. These terms simply mean that the hypertension is of unknown origin, in contrast to the forms of hypertension that are secondary to known causes, such as renal artery stenosis or mono- genic forms of hypertension.
In most patients, excess weight gain and a sedentary life- style appear to play a major role in causing primary hyper- tension. Most patients with hypertension are overweight, and studies of different populations have suggested that excess adiposity may account for as much as 65% to 75% of the risk for developing primary hypertension. Clinical studies have clearly shown the value of weight loss for reducing blood pressure in most patients with hyperten- sion, and clinical guidelines for treating hypertension rec- ommend increased physical activity and weight loss as a first step in treating most patients with hypertension.
The following characteristics of primary hyperten- sion, among others, are caused by excess weight gain and obesity:
1. Cardiac output is increased in part because of the additional blood flow required for the extra adipose tissue. However, blood flow in the heart, kidneys, gastrointestinal tract, and skeletal muscle also in- creases with weight gain because of increased met- abolic rate and growth of the organs and tissues in response to their increased metabolic demands. As the hypertension is sustained for many months and years, total peripheral vascular resistance may be
increased.
- Sympathetic nerve activity, especially in the kidneys, is increased in overweight patients. The causes of increased sympathetic activity in obese persons are not fully understood, but studies have suggested that hormones such as leptin that are released from fat cells may directly stimulate multiple regions of the hypothalamus, which in turn have an excitatory influence on the vasomotor centers of the brain me- dulla. There is also evidence for reduced sensitivity of arterial baroreceptors in buffering increases in arterial pressure, as well as activation of chemore- ceptors in obese persons, especially in those who also have obstructive sleep apnea.
- Angiotensin II and aldosterone levels are in- creased in many obese patients. This increase may be caused partly by increased sympathetic nerve stimulation, which increases renin release by the kidneys and therefore formation of angiotensin II, which in turn stimulates the adrenal gland to se- crete aldosterone.
- The renal-pressure natriuresis mechanism is im- paired, and the kidneys will not excrete adequate amounts of salt and water unless the arterial pres- sure is high or kidney function is somehow improved. If mean arterial pressure in the essential hyperten- sive person is 150 mm Hg, acute reduction of mean arterial pressure to the normal value of 100 mm Hg (but without otherwise altering renal function ex- cept for the decreased pressure) will cause almost total anuria. The person will then retain salt and water until the pressure rises back to the elevated value of 150 mm Hg. Chronic reductions in arterial pressure with effective antihypertensive therapies, however, usually do not cause marked salt and wa- ter retention by the kidneys because these therapies also improve renal pressure natriuresis, as discussed later.
Experimental studies in obese animals and obese patients have suggested that impaired renal pressure natriuresis in obesity hypertension is caused mainly by increased renal tubular reabsorption of salt and water due to increased sympathetic nerve activity and increased levels of angiotensin II and aldosterone. However, if hypertension is not effectively treated, there may also be vascular damage in the kidneys that can reduce glomeru- lar filtration rate and increase the severity of hyperten- sion. Eventually, uncontrolled hypertension associated with obesity and associated metabolic disorders can lead to severe vascular injury and complete loss of kidney function.
Graphic Analysis of Arterial Pressure Control in Essential Hypertension. Figure 19-15 is a graphic analysis of es- sential hypertension. The curves of this figure are called sodium-loading renal function curves because the arterial pressure in each case is increased very slowly, over many days or weeks, by gradually increasing the level of sodium intake. The sodium-loading type of curve can be deter- mined by increasing the level of sodium intake to a new level every few days and then waiting for the renal output of sodium to come into balance with the intake and, at the same time, recording the changes in arterial pressure.
When this procedure is used in patients with essen- tial hypertension, two types of curves, shown at the right in Figure 19-15, can be recorded; one is called (1) salt- insensitive hypertension and the other (2) salt-sensitive hypertension. Note in both cases that the curves are shifted to the right, to a higher pressure level than for people with normal arterial pressure. In the case of the person with salt-insensitive essential hypertension, the arterial pressure does not increase significantly when changing from a normal salt intake to a high salt intake. However, in patients who have salt-sensitive essential hypertension, the high salt intake significantly exacer- bates the hypertension.
Two additional points should be emphasized. First, salt sensitivity of blood pressure is not an all-or-none finding—it is quantitative, with some individuals being more salt-sensitive than others. Second, salt sensitivity of blood pressure is not a fixed characteristic; instead, blood pressure usually becomes more salt sensitive as a person ages, especially after 50 or 60 years of age, when the num- ber of functions units (nephrons) in the kidneys begins to decrease gradually.
The reason for the difference between salt-insensitive essential hypertension and salt-sensitive hypertension is presumably related to structural or functional differences in the kidneys of these two types of hypertensive patients. For example, salt-sensitive hypertension may occur with different types of chronic renal disease because of the gradual loss of nephrons or because of normal aging, as discussed in Chapter 32. Abnormal function of the renin-angiotensin system can also cause arterial pressure to become salt-sensitive, as discussed previously in this chapter.
Treatment of Essential Hypertension. As a first step, current guidelines for treating hypertension recommend lifestyle modifications aimed at increasing physical activ- ity and weight loss in most patients. Unfortunately, many patients are unable to lose weight, and pharmacological treatment with antihypertensive drugs must be initiated. Two general classes of drugs are used to treat hyperten- sion: (1) vasodilator drugs, which increase renal blood flow and glomerular filtration rate; and (2) natriuretic or diuretic drugs, which decrease tubular reabsorption of salt and water.
Vasodilator drugs usually cause vasodilation in many other tissues of the body, as well as in the kidneys. Dif- ferent vasodilators act in one of the following ways: (1) by inhibiting sympathetic nervous signals to the kidneys or by blocking the action of the sympathetic transmit- ter substance on the renal vasculature and renal tubules; (2) by directly relaxing the smooth muscle of the renal vasculature; or (3) by blocking the action of the renin- angiotensin-aldosterone system on the renal vasculature or renal tubules.
Drugs that reduce the reabsorption of salt and water by the renal tubules include, in particular, drugs that block active transport of sodium through the tubular wall; this blockage in turn also prevents the reabsorption of water, as explained earlier in the chapter. These natriuretic or diuretic drugs are discussed in greater detail in Chapter 32.
SUMMARY OF INTEGRATED MULTIFACETED SYSTEMS FOR ARTERIAL PRESSURE REGULATION
It is clear that arterial pressure is regulated not by a single pressure controlling system but instead by several inter- related systems, each of which performs a specific func- tion. For example, when a person bleeds so severely that the pressure falls suddenly, two problems confront the pressure control system. The first is survival; the arterial pressure must be rapidly returned to a high enough level that the person can live through the acute episode. The second is to return the blood volume and arterial pressure eventually to their normal levels so that the circulatory system can reestablish full normality, not merely back to the levels required for survival.
In Chapter 18, we saw that the first line of defense against acute changes in arterial pressure is the nervous control system. In this chapter, we have emphasized a second line of defense achieved mainly by kidney mecha- nisms for the long-term control of arterial pressure. How- ever, there are other pieces to the puzzle. Figure 19-16 helps put these pieces together.
Figure 19-16 shows the approximate immediate (sec- onds and minutes) and long-term (hours and days) con- trol responses, expressed as feedback gain, of eight arterial pressure control mechanisms. These mechanisms can be divided into three groups: (1) those that react rapidly, within seconds or minutes; (2) those that respond over an intermediate time period—that is, minutes or hours; and (3) those that provide long-term arterial pressure regula- tion for days, months, and years.
Arterial Pressure Control Mechanisms That Act With- in Seconds or Minutes. The rapidly acting pressure control mechanisms are almost entirely acute nervous reflexes or other autonomic nervous system responses. Note in Figure 19-16 the three mechanisms that show responses within seconds: (1) the baroreceptor feedback mechanism; (2) the central nervous system ischemic mechanism; and (3) the chemoreceptor mechanism. Not only do these mechanisms begin to react within seconds, but they are also powerful. After any acute fall in pressure, as might be caused by severe hemorrhage, the nervous mechanisms combine to cause the following: (1) constric- tion of the veins and transfer of blood into the heart; (2) increased heart rate and contractility of the heart to pro- vide greater pumping capability by the heart; and (3) con- striction of most peripheral arterioles. All these effects occur almost instantly to raise the arterial pressure back into a survival range.
When the pressure suddenly rises too high, as might occur in response to a rapid transfusion of excess blood, the same control mechanisms operate in the reverse direc- tion, again returning the pressure back toward normal.
Arterial Pressure Control Mechanisms That Act After Many Minutes. Several pressure control mechanisms exhibit significant responses only after a few minutes fol- lowing an acute arterial pressure change. Three of these mechanisms, shown in Figure 19-16, are as follows: (1) the renin-angiotensin vasoconstrictor mechanism; (2) stress relaxation of the vasculature; and (3) shift of fluid through the tissue capillary walls in and out of the circula- tion to readjust the blood volume as needed.
We have already described at length the role of the renin-angiotensin vasoconstrictor system to provide a semiacute means for increasing the arterial pressure when necessary. The stress relaxation mechanism is demonstrated by the following example. When the pres- sure in the blood vessels becomes too high, they become stretched more and more for minutes or hours; as a result, the pressure in the vessels falls toward normal. This con- tinuing stretch of the vessels, called stress relaxation, can serve as an intermediate-term pressure “buffer.”
The capillary fluid shift mechanism means simply that whenever capillary pressure falls too low, fluid is absorbed from the tissues through the capillary membranes and into the circulation, thus building up the blood volume and increasing the pressure in the circulation. Conversely, when the capillary pressure rises too high, fluid is lost out of the circulation into the tissues, thus reducing the blood volume, as well as virtually all the pressures throughout the circulation.
These three intermediate mechanisms mostly become activated within 30 minutes to several hours. During this time, the nervous mechanisms usually become less and less effective, illustrating the importance of these non- nervous, intermediate-term pressure control measures.
Long-Term Mechanisms for Arterial Pressure Regula- tion. The goal of this chapter has been to explain the role of the kidneys in long-term control of arterial pressure. To the far right in Figure 19-16 is shown the renal–blood volume pressure control mechanism, which is the same as the renal–body fluid pressure control mechanism, dem- onstrating that it takes a few hours to begin showing sig- nificant response. Yet, it eventually develops a feedback gain for control of arterial pressure that is nearly equal to infinity. This means that this mechanism can eventually return the arterial pressure nearly all the way back, not merely partway back, to the pressure level that provides normal output of salt and water by the kidneys.
Many factors can affect the pressure-regulating level of the renal–body fluid mechanism. One of these, shown in Figure 19-16, is aldosterone. A decrease in arterial pres- sure leads within minutes to an increase in aldosterone secretion and, over the next hour or days, this effect plays an important role in modifying the pressure control char- acteristics of the renal–body fluid mechanism.
Especially important is the interaction of the renin- angiotensin system with the aldosterone and renal fluid mechanisms. For example, a person’s salt intake varies tremendously from one day to another. We have seen in this chapter that salt intake can decrease to as little as one- tenth normal or can increase to 10 to 15 times normal and yet the regulated level of the mean arterial pressure will change only a few mm Hg if the renin-angiotensin- aldosterone system is fully operative. However, without a functional renin-angiotensin-aldosterone system, blood pressure becomes very sensitive to changes in salt intake.
Thus, arterial pressure control begins with the life- saving measures of the nervous pressure controls, then continues with the sustaining characteristics of the inter- mediate pressure controls and, finally, is stabilized at the long-term pressure level by the renal–body fluid mecha- nism. This long-term mechanism, in turn, has multiple interactions with the renin-angiotensin-aldosterone sys- tem, the nervous system, and several other factors that provide special blood pressure control capabilities for special purposes.

コメント