
Contents
- 1 ENZYMES: KINETICS (PART I)
- 1.1 エネルギーとは?
- 1.2 エネルギーの挙動は熱力学の法則によって支配される
- 1.3 熱力学に影響を与える要因
- 1.4 エントロピーとエンタルピーの関係
- 1.5 熱力学の4つの法則(Four Laws of Thermodynamics)
- 1.6 酵素動態(Enzyme Kinetics)
- 1.7 触媒反応と非触媒反応の違い
- 1.8 ギブス自由エネルギー(Gibbs Free Energy)
- 1.9 触媒された化学反応(Catalyzed Chemical Reactions)
- 1.10 酵素活性に影響を与える要因(Factors Affecting Enzyme Activity)
- 1.11 衝突理論(Kinetic Theory / Collision Theory)
- 1.12 温度(Temperature)と酵素反応速度
- 1.13 pHと酵素活性
- 1.14 基質濃度(Substrate Concentration, S)
- 2 ENZYMES: KINETICS (PART II) AND REGULATION
- 2.1 今日の学習目標(Learning Objectives)
- 2.2 用語の定義(Definition of Terms)
- 2.3 反応速度と速度定数(Rate Constant and Reaction Velocity)
- 2.4 平衡定数(Equilibrium Constant, Keq)
- 2.5 酵素動態における速度定数(Rate Constants)と基質濃度の影響
- 2.6 酵素動態における反応の次数(Reaction Orders in Enzyme Kinetics)
- 2.7 **アルコールデヒドロゲナーゼ(Alcohol Dehydrogenase, ADH)**と零次反応
- 2.8 一次反応動態(First-Order Kinetics)
- 2.9 二次反応動態(Second-Order Kinetics)
- 2.10 まとめ:反応次数と基質濃度に関する反応速度(Reaction Orders with Respect to Substrate Concentration)
- 2.11 Michaelis-Menten式と酵素動態の関係
- 2.12 Lineweaver-Burkeプロット
- 2.13 Dixonプロット
- 2.14 酵素阻害の種類(Types of Enzyme Inhibition)
- 2.15 競争的阻害(Competitive Inhibition)
- 2.16 非競争的阻害(Noncompetitive Inhibition)/混合型阻害(Mixed Inhibition)
- 2.17 反競争的阻害(Uncompetitive Inhibition)
- 2.18 阻害タイプごとのKmとVmaxへの影響
- 2.19 なぜ酵素活性を調節するのか?(Why Regulate Enzyme Activity?)
- 2.20 コンパートメンタリゼーション(Compartmentalization)
- 2.21 MECHANISMS FOR ENZYME REGULATION
- 2.22 酵素の調節メカニズム(Mechanisms for Enzyme Regulation)
- 2.23 遅い制御(Slow Control)
- 2.24 酵素合成の制御(Control of Enzyme Synthesis)
- 2.25 酵素分解の制御(Control of Enzyme Degradation)
- 2.26 迅速な制御(Rapid Control)
- 2.27 アロステリック調節(Allosteric Regulation)
- 2.28 Hill式と協同結合(Hill Equation and Cooperative Binding)
- 2.29 アロステリック調節とシグモイド曲線(Allosteric Regulation and Sigmoidal Curve)
- 2.30 共有結合修飾(Covalent Modification)
- 2.31 プロテオリティック活性化(Proteolytic Activation)
- 2.32 フィードバック調節(Feedback Regulation)
- 2.33 今日の学習目標(Learning Objectives for Today)
ENZYMES: KINETICS (PART I)
エネルギーとは?
エネルギー(Energy)とは、物体が仕事(Work)をする能力のことです。エネルギーには主に二つの形があります:
- 位置エネルギー(Potential energy): 物体が持つ潜在的なエネルギー。たとえば、原子内部のエネルギーや重力エネルギーが該当します。
- 運動エネルギー(Kinetic energy): 物体が動いている時に持つエネルギー。熱エネルギー(Thermal energy)もこの一部です。

エネルギーの挙動は熱力学の法則によって支配される
**熱力学(Thermodynamics)**は、エネルギーとその変換に関する物理学の一分野です。特に、熱(Heat)や温度(Temperature)がエネルギーや仕事に与える影響を研究します。
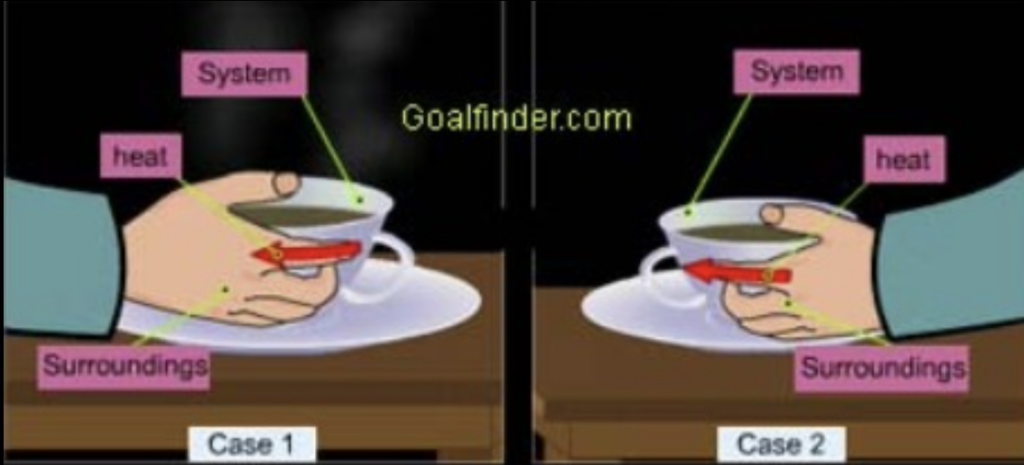
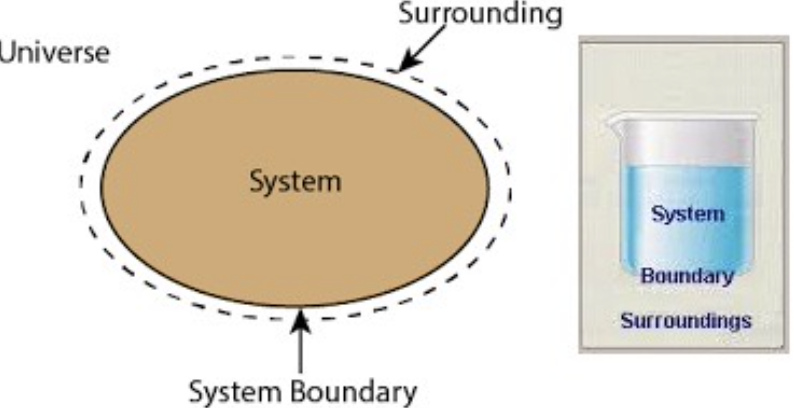
- システム(System): 研究対象となる領域で、境界(Boundary)によって囲まれています。
- 外部環境(Surrounding): システムの外部領域です。
- 宇宙(Universe): システムと外部環境を合わせたものです。
熱力学に影響を与える要因
- エンタルピー(Enthalpy, H): システム内のエネルギーの総量を表します。エンタルピーは内部エネルギー(Internal Energy, U)と圧力(Pressure, P)、体積(Volume, V)の合計です。
- H = U + PV
- エンタルピー変化(ΔH): システムが閉じている場合、エンタルピーの変化はシステム内の内部エネルギーの変化を表します。
- 正のΔH: システムがエネルギーを吸収したことを示します。(例:吸熱反応、Endothermic Reaction)
- 負のΔH: システムがエネルギーを放出したことを示します。(例:発熱反応、Exothermic Reaction)
- ΔH = 0 はエネルギーの吸収・放出がないことを示します。


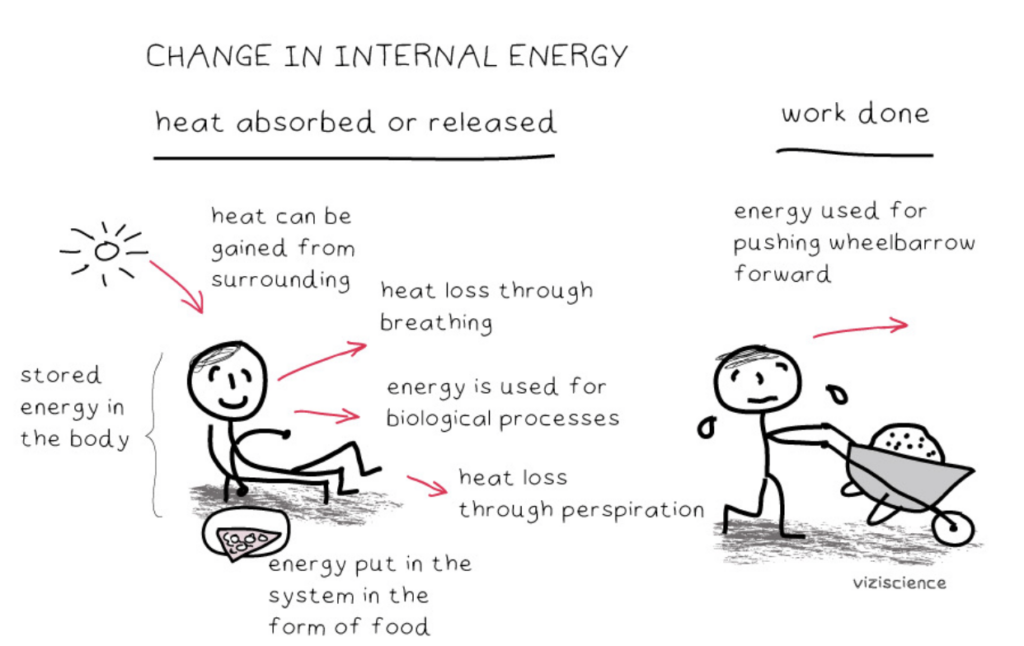
- エントロピー(Entropy, S): 分子の無秩序やランダムさの度合いを表します。エントロピーはシステム内で有効に利用されなかったエネルギーの量も示します。
エントロピーとエンタルピーの関係
熱力学的なシステムでは、エンタルピー変化(ΔH)とエントロピー変化(ΔS)の組み合わせが、反応がどのように進行するかを決定します。
| 反応の種類 | エネルギーの変化 | ΔH(エンタルピー変化) | 生成物と反応物のエネルギー関係 |
|---|---|---|---|
| 発熱反応(Exothermic Reaction) | エネルギーが周囲に放出される | ΔHは負(negative) | 生成物のエネルギーは反応物より少ない |
| 吸熱反応(Endothermic Reaction) | エネルギーが周囲から吸収される | ΔHは正(positive) | 生成物のエネルギーは反応物より多い |
状況別の例
| ΔH | ΔS | 反応の進行 |
|---|---|---|
| 正(Positive) | 正(Positive) | 高温で自発的に進行する(エントロピーが増加するため、TΔSの影響が大きい) |
| 負(Negative) | 負(Negative) | 低温で自発的に進行する(エントロピーの減少は不利だが、ΔHが負であるため有利) |
| 正(Positive) | 負(Negative) | 反応は非自発的(エントロピーが減少し、エンタルピーも増加するため不利) |
| 負(Negative) | 正(Positive) | 反応は自発的(エンタルピーが減少し、エントロピーも増加するため有利) |
解説
- ΔHが正かつΔSが正: 高温で反応が自発的に進行します。エネルギーを吸収しながら、エントロピーが増加するため、温度が高いほどTΔSが反応を有利にします。
- ΔHが負かつΔSが負: 低温で反応が自発的に進行します。エネルギーを放出しながら、エントロピーが減少するため、温度が低い方が有利です。
- ΔHが正かつΔSが負: 反応は非自発的です。エネルギーを吸収し、エントロピーも減少するため、どの温度でも不利な反応です。
- ΔHが負かつΔSが正: 反応は自発的に進行します。エネルギーを放出し、エントロピーが増加するため、反応が非常に有利です。
熱力学の4つの法則(Four Laws of Thermodynamics)
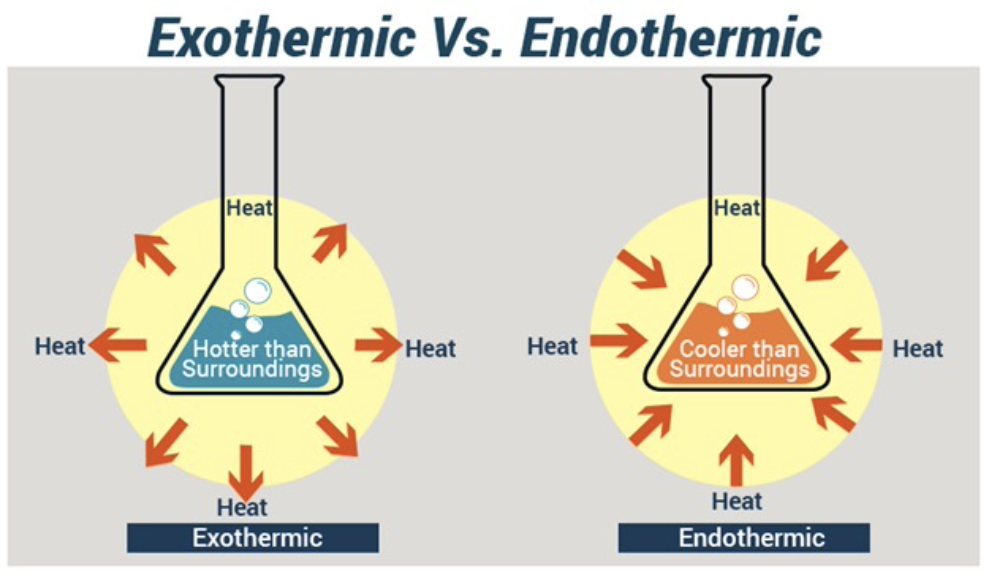
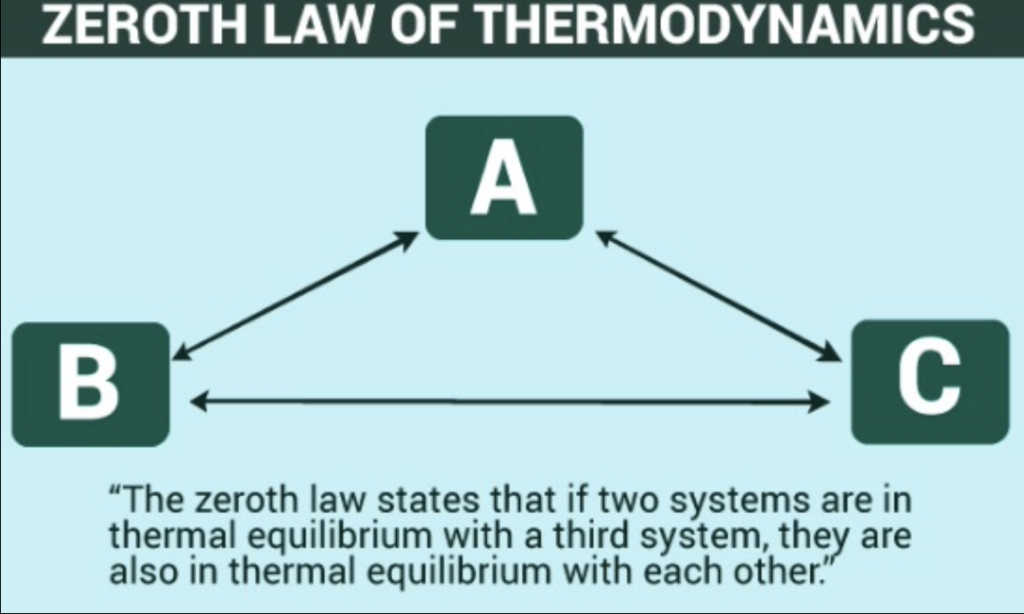
第零法則(Zeroth Law of Thermodynamics)
- 2つの物体が熱的平衡状態(Thermal Equilibrium)にある場合、それらの温度は同じです。
- もし2つの物体が「接触」しており、間に熱の流れがない場合、これらは熱的平衡にあります。
第一法則(First Law of Thermodynamics)
- 閉じたシステムの内部エネルギーの変化は、システムに供給された熱量からシステムが行った仕事を差し引いたものです。
- エネルギー保存の法則(Law of Conservation of Energy): 孤立したシステムの全エネルギーは時間とともに一定であり、エネルギーは形を変えることができるが、創造も破壊もされません。
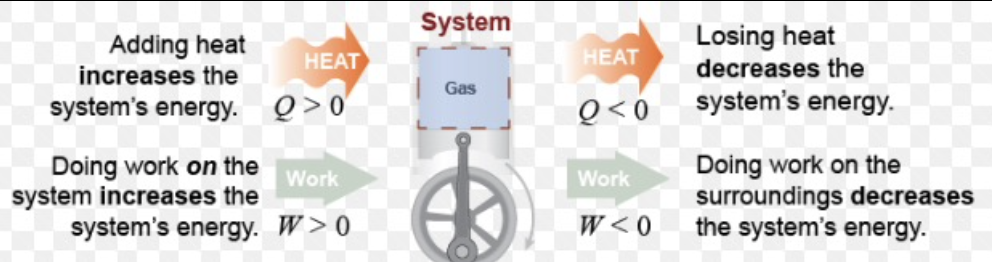

第二法則(Second Law of Thermodynamics)
- 孤立したシステムのエントロピー(Entropy)は常に増加します。孤立系は自発的に熱平衡に向かい、エントロピーが最大になります。
- 宇宙全体のエントロピーは常に増加し、決して減少しません。この現象は「エントロピーの増大の法則(Law of Increased Entropy)」と呼ばれ、不可避の劣化や崩壊を意味します。
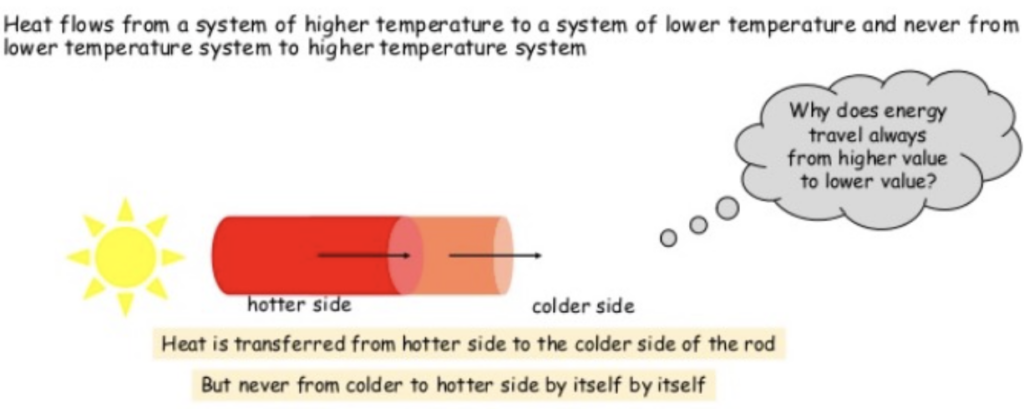
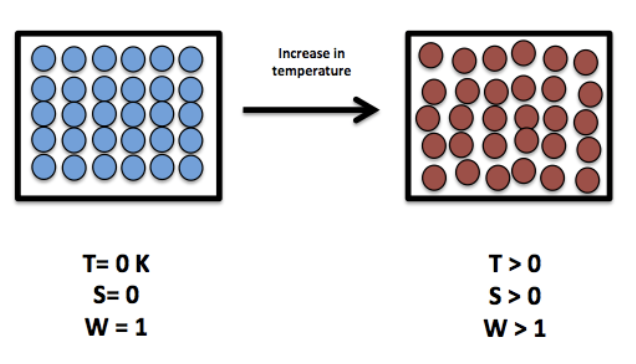
第三法則(Third Law of Thermodynamics)
- 物体の温度が絶対零度(Absolute Zero)に近づくと、そのエントロピーは一定の値に近づきます。
酵素動態(Enzyme Kinetics)
酵素動態とは?
- すべての生物に共通する基本的な特性として、低温でも何千もの化学反応が起こっています。これらの反応は非常に高い速度で、かつ高度に調整された形で行われており、酵素(Enzyme)の助けを借りています。
- 酵素動態は、酵素が触媒する反応の速度(Rate of Enzyme-Catalyzed Reactions)と、その速度に影響を与えるすべての要因を研究する分野です。これには以下が含まれます:
- 基質濃度(Substrate Concentration)
- pH
- 温度(Temperature)
- イオン強度(Ionic Strength)
触媒反応と非触媒反応の違い
非触媒反応(Uncatalyzed Reactions / Spontaneous Reactions)
- 基質(Substrate): 例として水素(H₂)と酸素(O₂)の分子は、外部から力が加わることなく衝突し、反応が自発的に進行します。
- 自発的な反応は、システムの**エンタルピー(Enthalpy)**が減少し、**エントロピー(Entropy)**が増加するときに起こりやすいです。
自発的/自然なプロセスの例
- 部屋で香りが拡散する
- 滑り台でボールが転がる
- 爆弾が加熱されて爆発する
- ぬるま湯の中で氷が溶ける
- 水に塩が溶ける
- 鉄がさびる
自発的反応で何が起こるか?
- エンタルピー(ΔH): 自発的な反応では、エンタルピーは減少します(システムが熱を放出する)。
- エントロピー(ΔS): エントロピーは増加します(システム内の無秩序が増加する)。
- 温度(Temperature): 温度が増加すると、反応が進行しやすくなります。
自発的プロセスのパラメータ
| パラメータ | 自発的反応(Spontaneous) | 非自発的反応(Nonspontaneous) |
|---|---|---|
| ΔH | 減少 | 増加 |
| ΔS | 増加 | 減少 |
| 温度 | 増加 | 減少 |
ギブス自由エネルギー(Gibbs Free Energy)
- ギブス自由エネルギー(Gibbs Available Energy): システム内で実際に仕事をすることができるエネルギーの量を示す式です。
- 反応が進む方向(自発的か非自発的か)や、平衡状態における反応物と生成物の濃度を説明します。
ギブス自由エネルギーの式
- ΔG = ΔH – TΔS
- ΔG: ギブス自由エネルギー変化
- ΔH: エンタルピー変化
- T: 絶対温度(Kelvin単位)
- ΔS: エントロピー変化
パラメータと反応の自発性
| パラメータ | 自発的(Spontaneous) | 非自発的(Nonspontaneous) |
|---|---|---|
| ΔH | 減少 | 増加 |
| ΔS | 増加 | 減少 |
| 温度 | 増加 | 減少 |
ギブス自由エネルギーと反応の関係
- ΔG < 0: 反応は自発的です(反応が進行する)。
- ΔG > 0: 反応は非自発的です(反応が進行しない)。
- ΔG = 0: 反応は平衡状態にあります。
触媒された化学反応(Catalyzed Chemical Reactions)
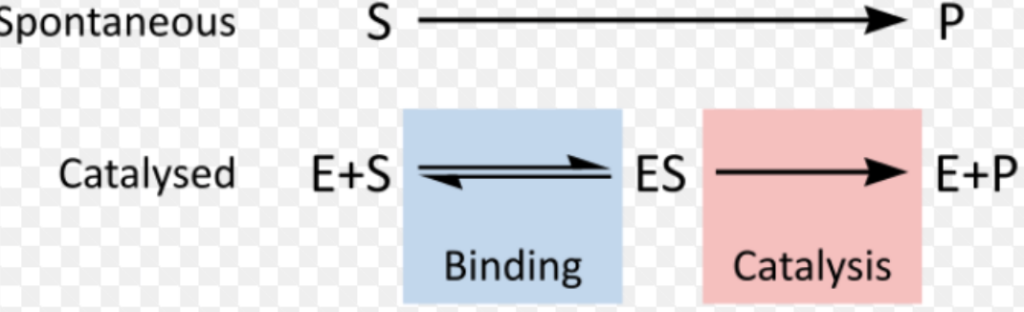
酵素触媒反応におけるギブス自由エネルギー(Gibbs Free Energy of Enzyme-Catalyzed Reactions)
- **ギブス自由エネルギー(Gibbs Free Energy)**は、生成物(Products)の形成に使用されるエネルギーの合計から、酵素-基質複合体(ES Complex)の形成に使用されるエネルギーを差し引いたものです。
- ギブス自由エネルギーは、反応の進行方向(ΔG)と、平衡時の反応物と生成物の濃度(平衡定数(Keq))を示します。
重要なパラメータ
- ΔG0: 標準状態での利用可能エネルギー(pH 7、温度37°C、一定圧力の条件下)。
- ΔG0’: 非標準状態での利用可能エネルギー(ΔGp – ΔGs、生成物と基質の自由エネルギー差)。
- Keq: 平衡時の生成物と基質の濃度比。Keq = [P] / [S]。
ΔG0を負にするための条件
- **エントロピー(Entropy)**を増加させる必要があります。
- 生成物の濃度が反応物の濃度よりも高くなる必要があります。
- Keq=[P]/[S]
ΔGと反応の関係
| パラメータ | 自発的反応(Spontaneous) | 非自発的反応(Nonspontaneous) |
|---|---|---|
| ΔG0 | 負 | 正 |
| Keq | 増加 | 減少 |
| ΔG0′ | 負 | 正 |
ギブス自由エネルギーの特徴
- ギブス自由エネルギーは、反応の初期状態と最終状態についての情報を提供します。
- 化学反応のメカニズムには依存しないため、反応速度についての情報は提供しません。
- ギブス自由エネルギーは次の2つの点を示します:
- 反応の方向(自発的か非自発的か)
- 反応物と生成物の平衡状態
酵素活性に影響を与える要因(Factors Affecting Enzyme Activity)
- 温度(Temperature)
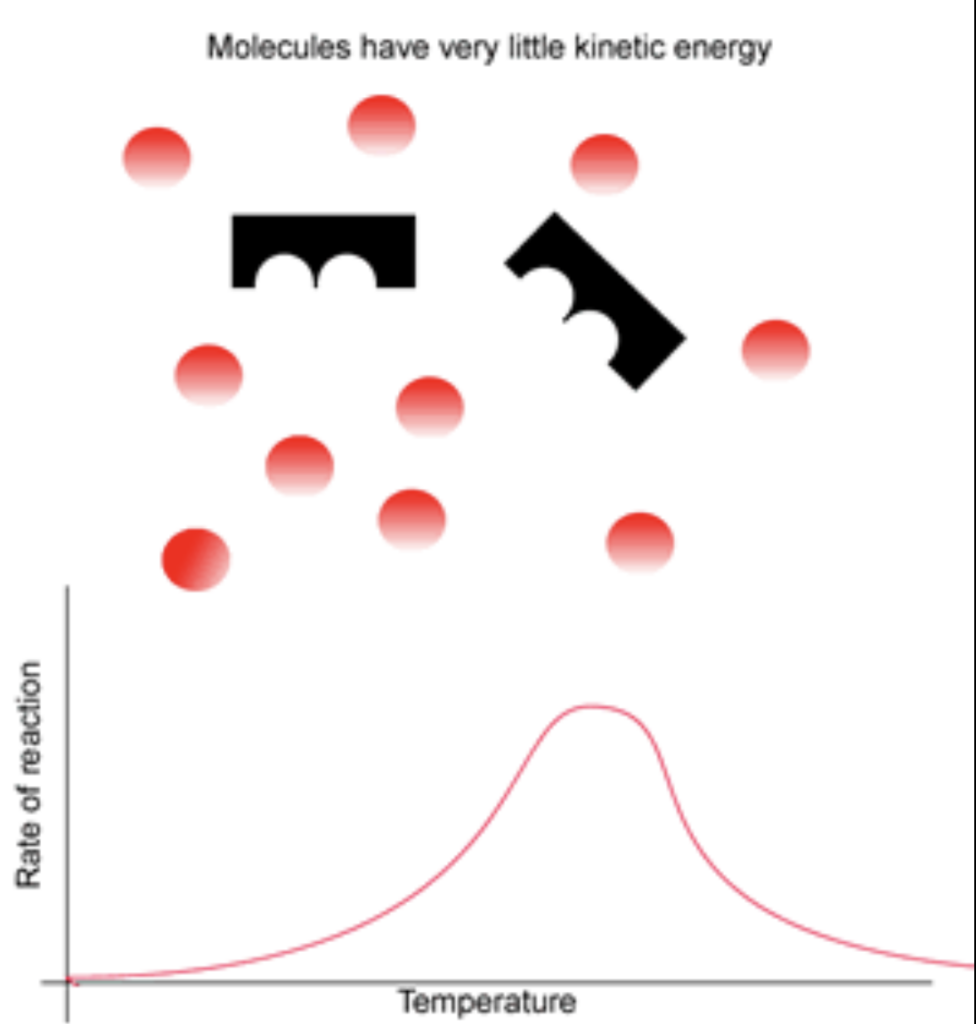
- 反応速度は温度が上昇するにつれて増加します。
- 温度が上昇すると、分子の運動エネルギーが増加し、衝突の頻度が高まります。
- 温度が10°C上昇すると、多くの酵素反応の活性は50〜100%増加します。
- 酵素が最も効率的に働く最適温度は37°Cです。
- 高温(45–55°C)で酵素は安定性を示し、65°C以上では不可逆的な失活が起こります。
- pH
- 各酵素反応には最適pHがあります。最適pHは常に生理的pHとは限りません。
- 細胞内酵素はpH 5〜9で最適活性を示します。
- 酵素による酸-塩基触媒作用に影響を与えます。
- 基質濃度(Substrate Concentration)
- 基質分子の濃度が増加すると、分子間の衝突の頻度が増加します。
- 例えば、基質AとBが反応してABを形成する場合、Aの濃度を2倍にすると衝突頻度が2倍に、AとBの両方の濃度を2倍にすると衝突頻度は4倍に増加します。
衝突理論(Kinetic Theory / Collision Theory)
- 2つの分子が反応するためには、以下の条件を満たす必要があります:
- 結合形成が可能な距離まで近づくこと。
- 反応が進行するために必要な活性化エネルギー(Activation Energy)を超える十分な運動エネルギーを持つこと。
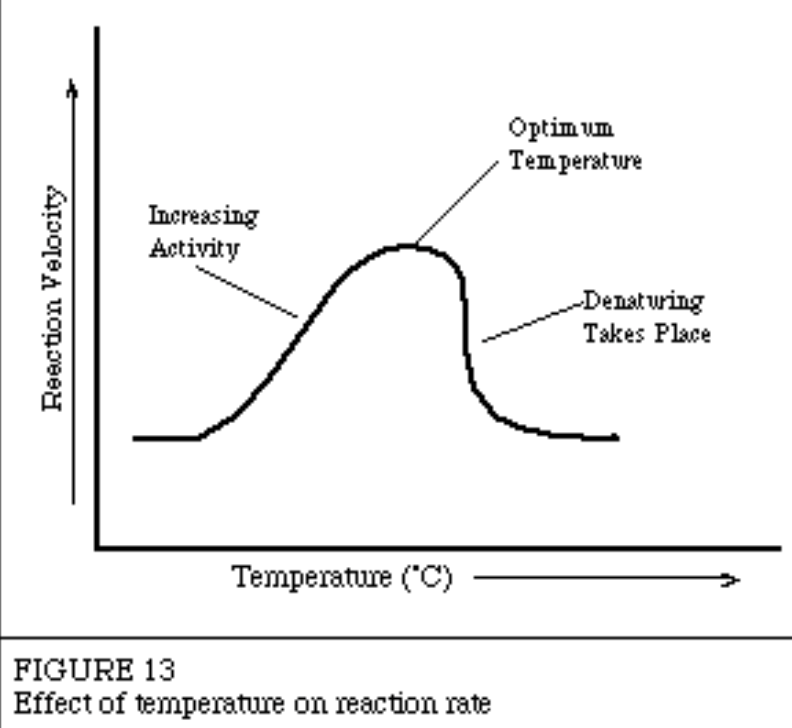
温度(Temperature)と酵素反応速度
- 反応速度は温度が上昇するにつれて増加します。
- 理由: 温度の上昇は分子の運動エネルギーを増加させ、分子間の衝突頻度が高くなるためです。
- 温度が10°C上昇すると、ほとんどの酵素活性は50〜100%増加します。
- 酵素が最もよく働く最適温度は37°Cです。
- 酵素は高温(45–55°C)ではまだ安定していますが、65°C以上では酵素の不可逆的な失活(変性)が起こります。
- 酵素はタンパク質分子なので、過度な高温によって構造が崩れ、機能を失います(変性, Denaturation)。
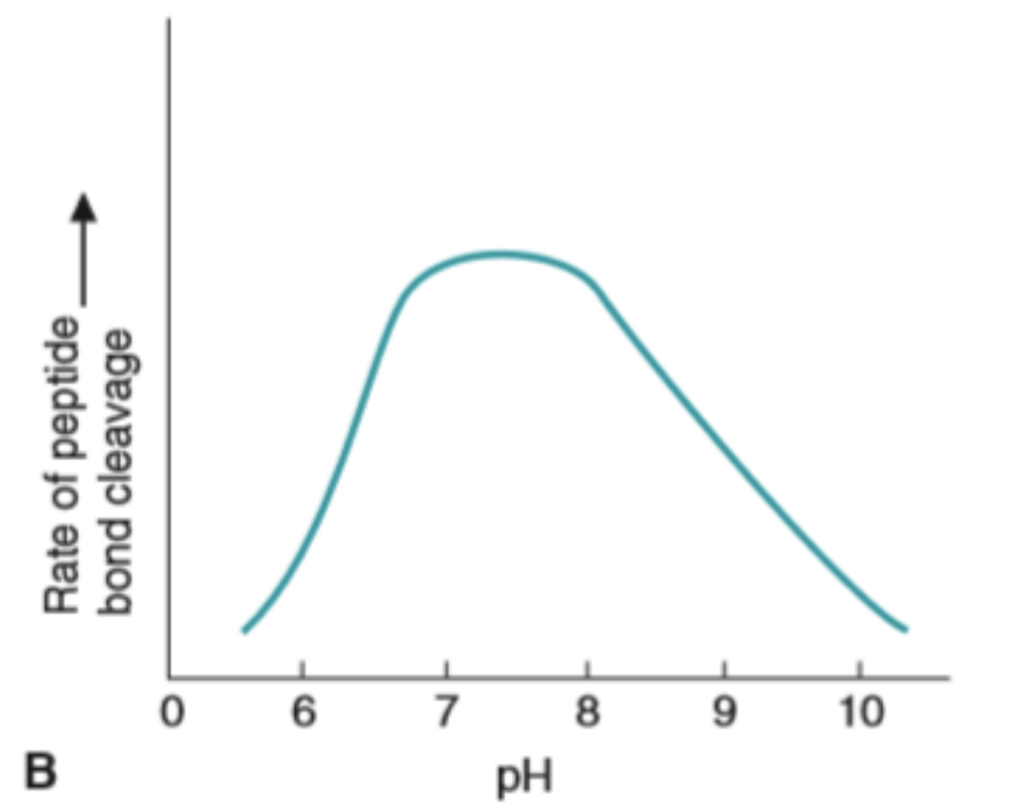
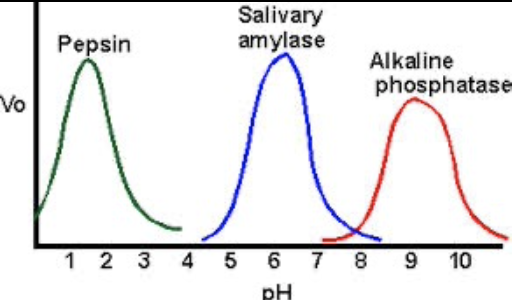
pHと酵素活性
- 酵素が触媒する反応には、各々に最適pHが存在します。最適pHは必ずしも生理的pH(pH 7.4)とは限りません。
- 細胞内酵素(Intracellular Enzymes)は、pH 5〜9の範囲で最適な活性を示します。
- 酸-塩基触媒(Acid-Base Catalysis)も酵素反応において重要な役割を果たします。適切なpHは、この触媒機構に影響を与えます。
基質濃度(Substrate Concentration, S)
- 基質分子の濃度が多ければ多いほど、分子間の衝突頻度が増加します。
- 例えば、反応式が A + B → AB で表される場合、基質Aの濃度を2倍にすると、衝突頻度は2倍になります。
- AとBの両方の濃度を2倍にすると、衝突頻度は4倍に増加します。
ENZYMES: KINETICS (PART II) AND REGULATION
今日の学習目標(Learning Objectives)
- **Michaelis-Menten式(Michaelis-Menten Equation)とHill式(Hill Equation)**を説明し、その用途を理解する
- 競争的阻害(Competitive Inhibition)、非競争的阻害(Noncompetitive Inhibition)、**反競争的阻害(Uncompetitive Inhibition)**のメカニズムを理解し説明する
- Lineweaver-Burkeプロットの説明とその用途を理解する
- 多くの酵素における細胞内基質濃度がKmに近い理由を説明する
- 酵素の調節方法(Enzyme Regulation)を列挙し、それぞれを説明する
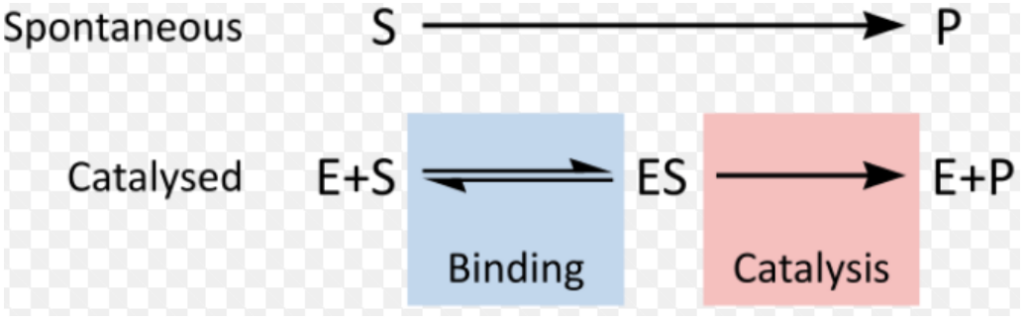
用語の定義(Definition of Terms)
ギブス自由エネルギー(Gibbs Free Energy)
- **閉じたシステム(Closed System)**において、仕事をするために使用可能なエネルギー。
反応速度(Reaction Velocity)
- 化学反応の速度を示します。これは、基質の濃度変化や生成物の濃度変化を時間で測ったものです。
- 式: V = Δ[基質または生成物の濃度] / 時間
速度定数(Rate Constant, k)
- 特定の反応における比例定数です。反応速度は基質濃度と速度定数に依存します。
- 式: 反応速度 = k [A][B]
反応速度と速度定数(Rate Constant and Reaction Velocity)
- k₁: 酵素が基質と結合して**酵素-基質複合体(Enzyme-Substrate Complex, ES Complex)**を形成する反応の速度定数。
- k₂: 触媒作用が行われ、最終的に反応生成物を生成し、酵素が再生される過程。このステップが**律速段階(Rate Limiting Step)**となる。
- k₃: 酵素-基質複合体が解離して、自由な酵素と基質に戻る反応の速度定数。
- k₄: 触媒反応の逆反応の速度定数。
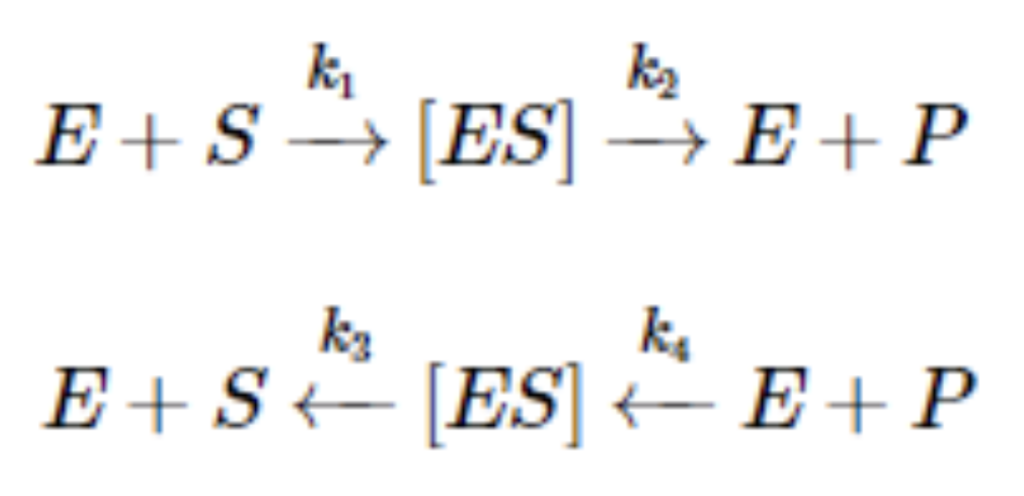
平衡定数(Equilibrium Constant, Keq)
- 反応が平衡状態にあるときの生成物と基質の濃度比。
- 式: Keq = [生成物] / [基質]
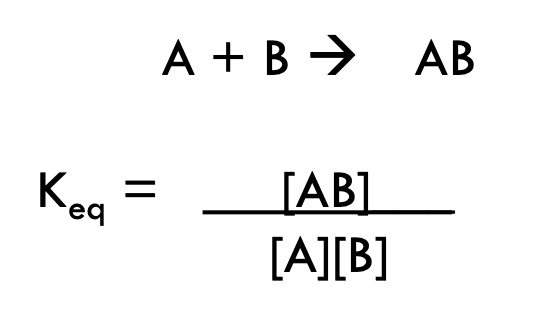
Keqの値と反応の傾向
- Keq = 0: 基質と生成物の濃度が等しい。
- Keq > 1: 生成物の生成が優位になる反応。
- Keq < 1: 基質の存在が優位になる反応。
/hr No matter how much increase in alcohol is present, ADH enzyme will only be able to catalyze 0.015g/100mL/hr Excessive amounts of alcohol intake will lead to accumulation
酵素動態における速度定数(Rate Constants)と基質濃度の影響

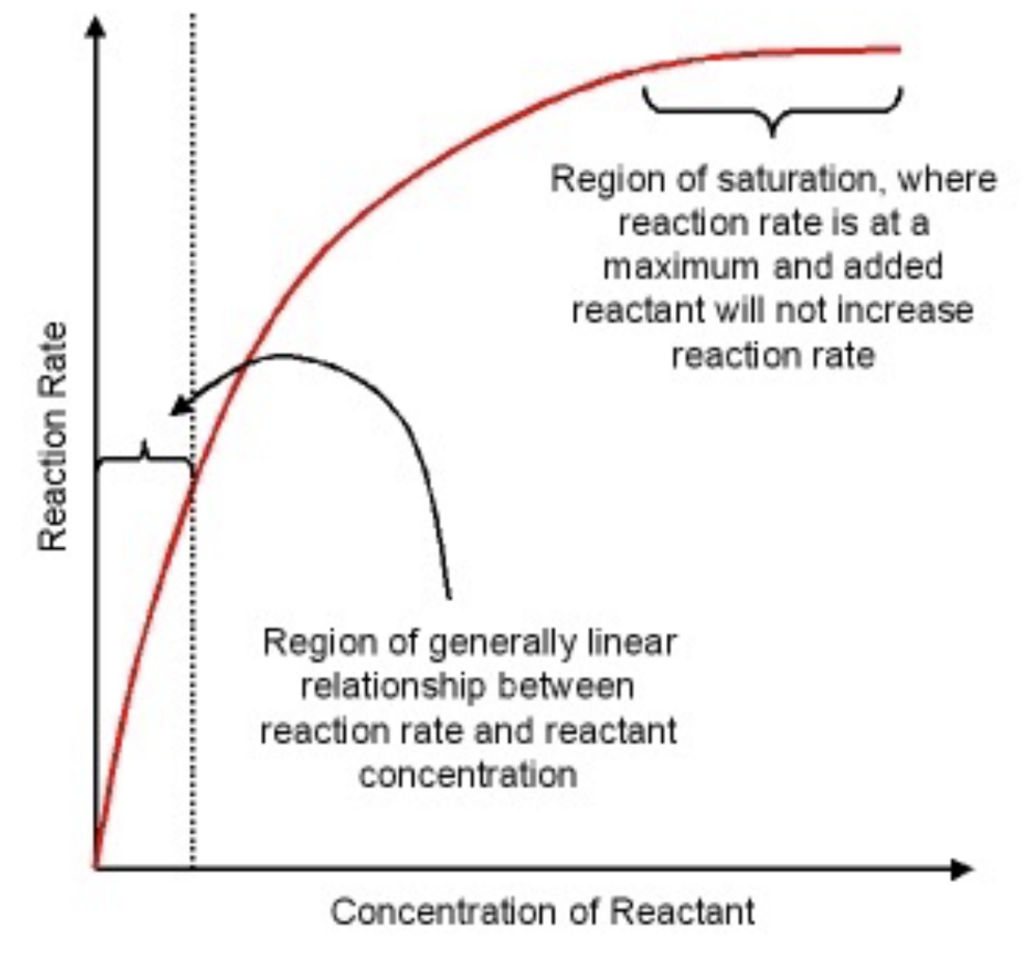
基質濃度と反応速度の関係
- **典型的な酵素(Typical Enzyme)**では、基質濃度(Substrate Concentration)を増加させると、初速度(Initial Velocity, Vi)が増加します。しかし、ある点に達すると、反応速度は最大値(Vmax)に達し、それ以上の基質濃度の増加では反応速度が上昇しません。
- この状態は、酵素が基質に「飽和(Saturated)」した状態を意味します。すべての酵素が基質で占有され、追加の基質を処理できない状況です。
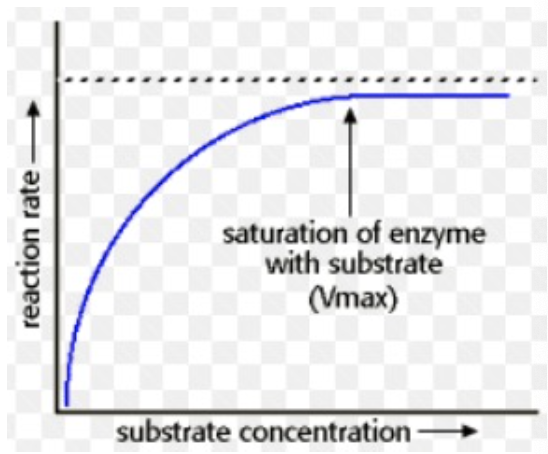
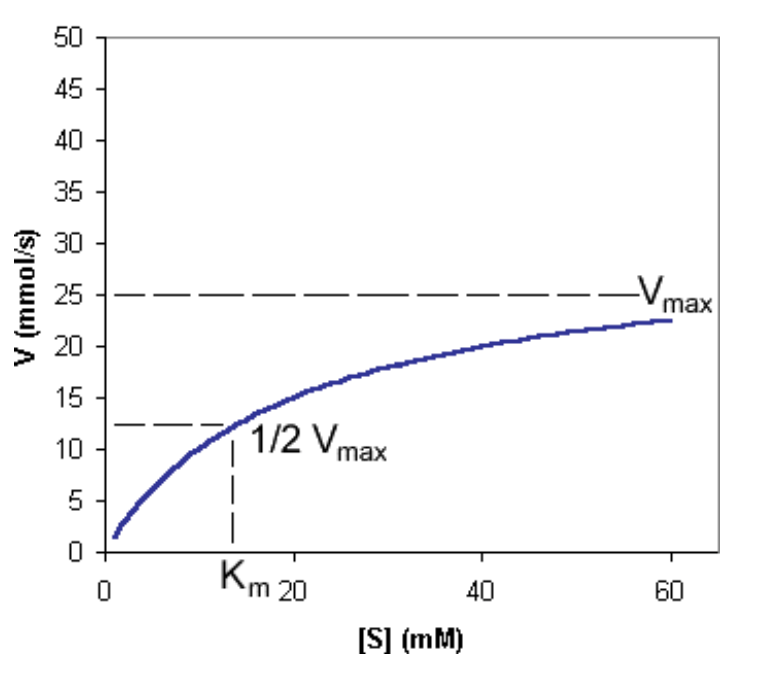
Vmax(最大反応速度)
- Vmaxは反応速度の最大値で、すべての酵素が完全に基質と結合し、追加の基質を処理できない状態を指します。
Km(ミカエリス定数)
- Kmは、反応速度が最大速度の50%に達するために必要な基質濃度を示します。Kmは酵素の基質に対する親和性を表しており、Kmが低いほど酵素は基質に対して高い親和性を持ちます。
酵素動態における反応の次数(Reaction Orders in Enzyme Kinetics)
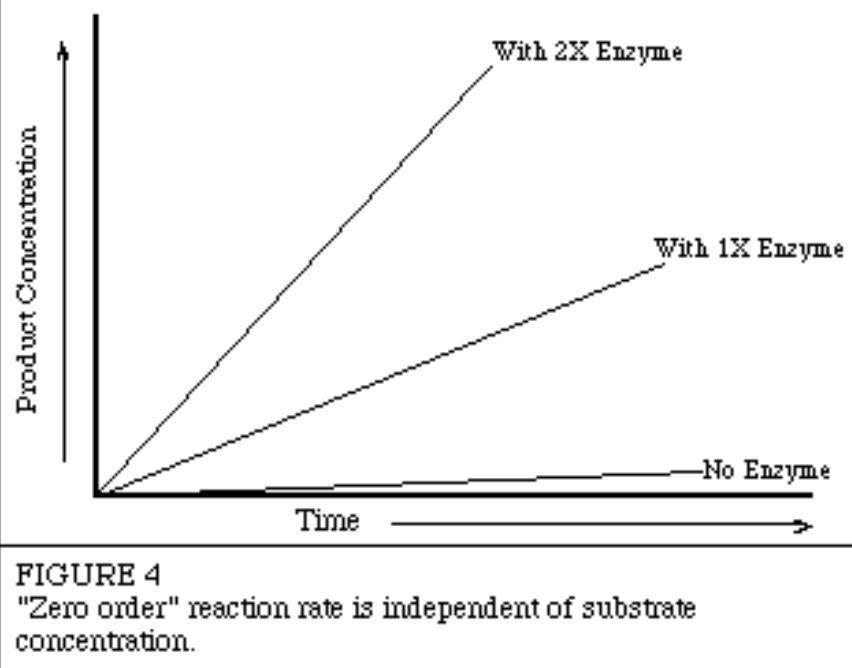
- 零次反応動態(Zero Order Kinetics)
- 酵素反応の速度はkに等しく、基質濃度に依存しません。すなわち、基質濃度が増加しても、反応速度は変わりません。
- 式: V = k
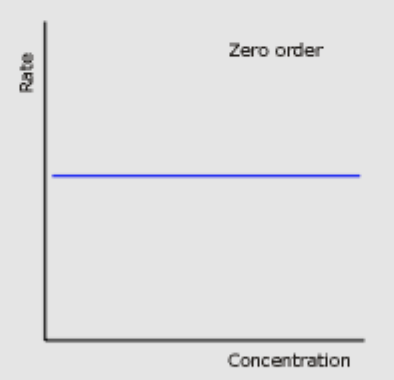
- この場合、生成される生成物の量は、存在する酵素のレベルに依存します。
- 一次反応動態(First Order Kinetics)
- 反応速度は基質濃度に比例します。
- 二次反応動態(Second Order Kinetics)
- 反応速度は2つの反応物の濃度の積に比例します。
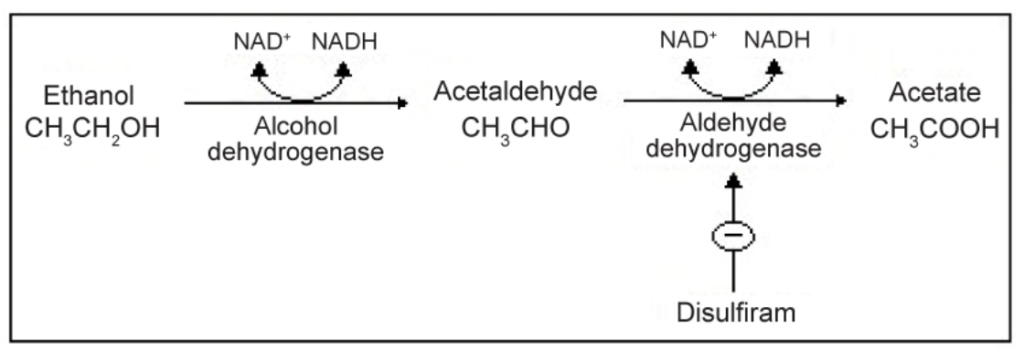
**アルコールデヒドロゲナーゼ(Alcohol Dehydrogenase, ADH)**と零次反応
- **アルコールデヒドロゲナーゼ(ADH)**は、エタノール(アルコール)をアセトアルデヒドに酸化する酵素です。この反応では、NAD⁺がNADHに還元されます。
- ADHは、エタノールを毎時0.015g/100mLの速度でアセトアルデヒドに変換します。
- 零次反応の典型例として、どれだけエタノールが増加しても、ADHが触媒する速度は0.015g/100mL/時に固定されます。
- したがって、過剰なアルコール摂取は体内にエタノールが蓄積し、酔いの原因となります。
一次反応動態(First-Order Kinetics)
- 反応速度は基質濃度に比例します。つまり、基質濃度が増加すると反応速度も増加します。多くの酵素はこの動態に従います。
二次反応動態(Second-Order Kinetics)
- 反応速度は2つの反応物の濃度または、1つの反応物の濃度の二乗に依存します。2つの反応物の濃度が関与するか、1つの反応物の濃度が二乗されることで反応が進行します。
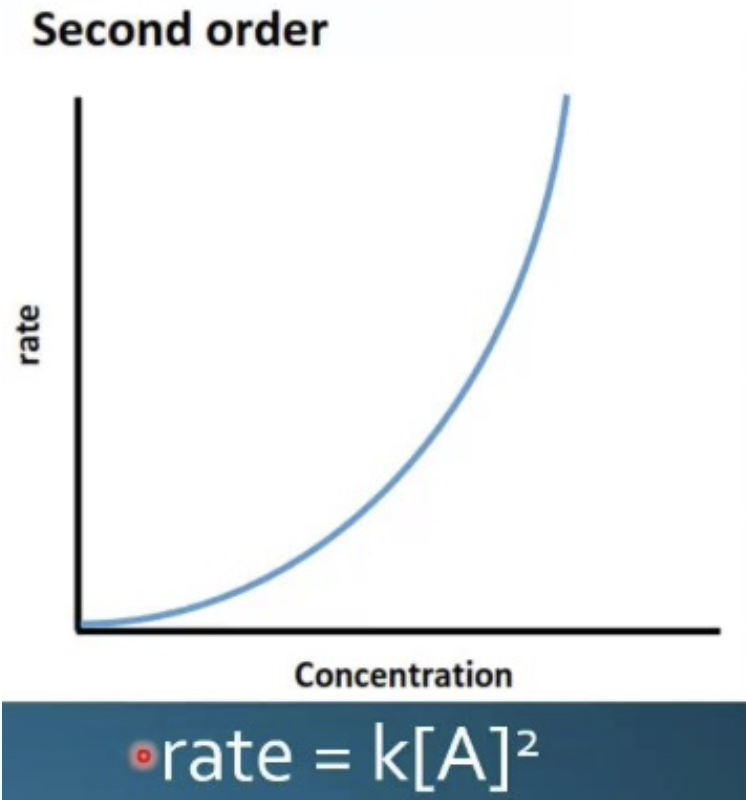
まとめ:反応次数と基質濃度に関する反応速度(Reaction Orders with Respect to Substrate Concentration)
| 反応次数 | 速度式 | コメント |
|---|---|---|
| 零次反応 | rate = k | 基質濃度に依存しない |
| 一次反応 | rate = k[S] | 基質濃度に比例する |
| 二次反応 | rate = k[S][S] または k[S]² | 基質濃度の二乗に比例する |
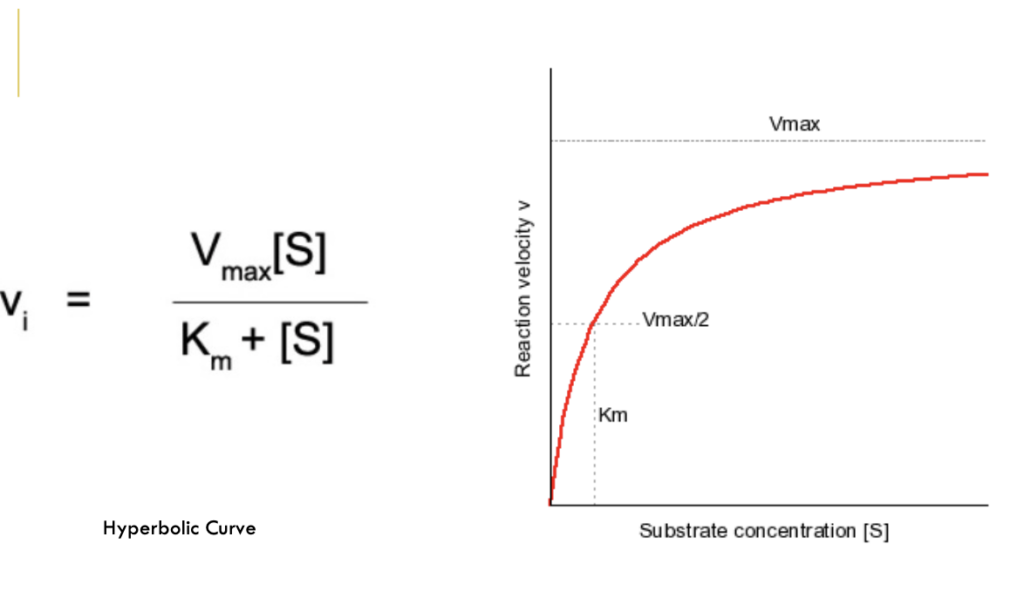
Michaelis-Menten式と酵素動態の関係
Michaelis-Menten式(Michaelis-Menten Equation)
- **レオノール・ミカエリス(Leonor Michaelis)とモード・メンテン(Maud Menten)**によって名付けられた式です。この式は、酵素反応速度と基質濃度の関係を表しています。
- ハイパボリック曲線(Hyperbolic Curve): Michaelis-Menten式は、基質濃度が増加するにつれて反応速度が飽和する現象を説明します。
定常状態仮定(Steady-State Assumption)
- 酵素-基質複合体(ES Complex)の濃度が一定となる点です。これは、ESの形成速度が解離速度と等しいことを意味します。

ミカエリス定数(KM, Michaelis Constant)
- 酵素が定常状態(Steady State)にあるときの基質濃度を表す定数です。Kmの値は多くの酵素において、細胞内の基質濃度に近い値となり、容易な調節が可能になります。
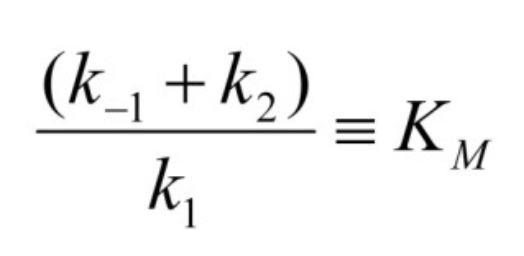
Kmの意義
- Kmが小さい酵素は、低い基質濃度でも最大の触媒効率に達します。したがって、Kmの値が小さいほど酵素の効率は高いです。
- Kmの値は基質、pH、温度により影響を受けます。
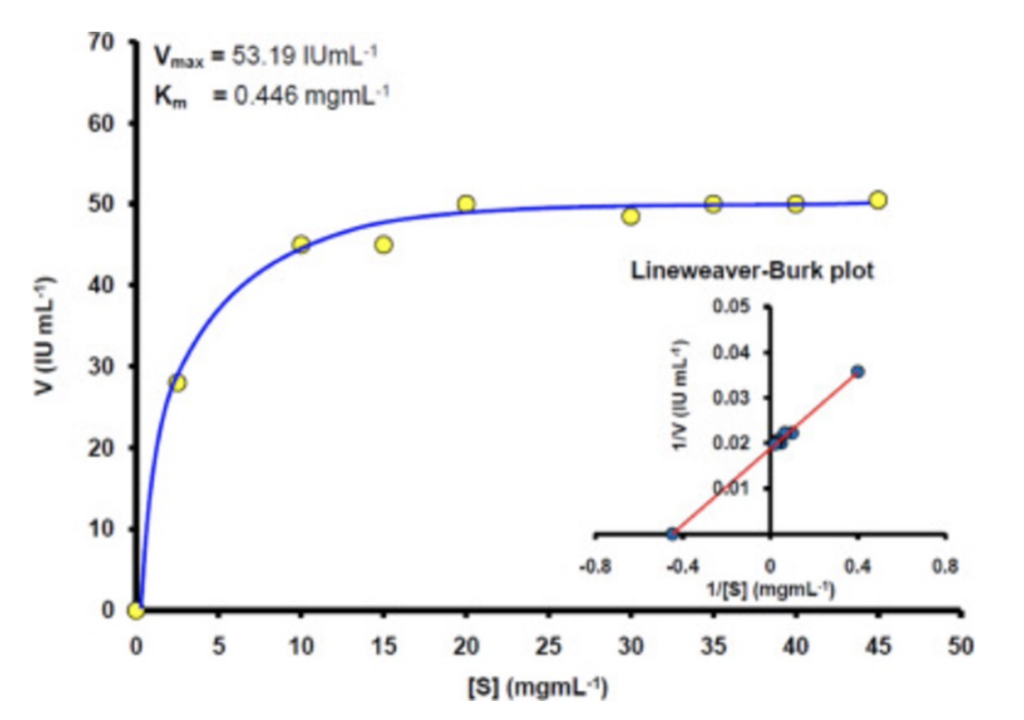
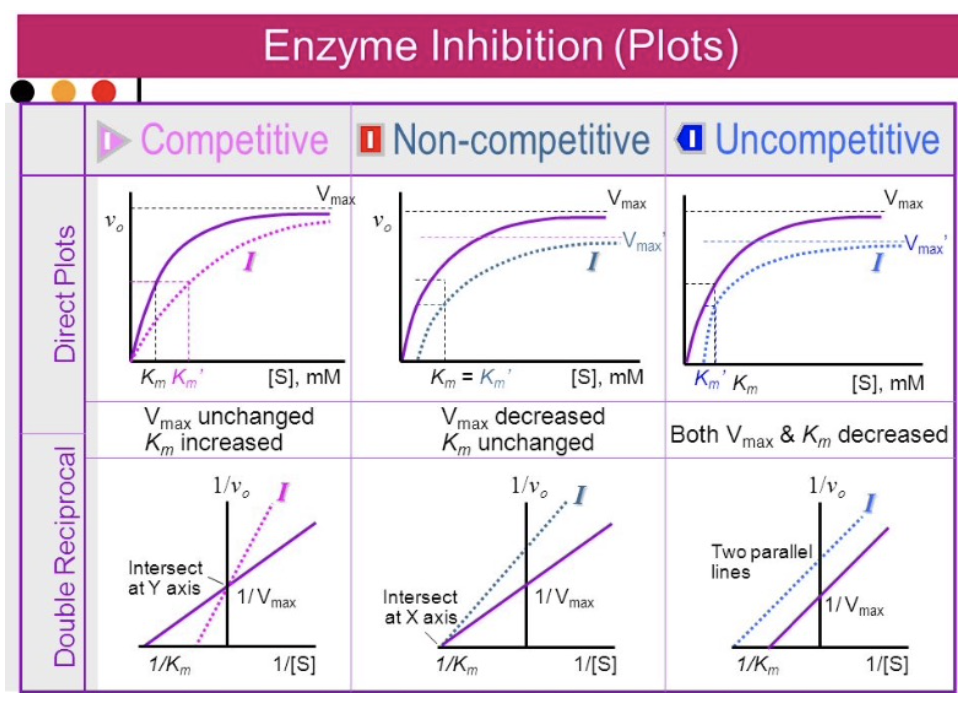
Lineweaver-Burkeプロット
- Lineweaver-Burkeプロットは、初速度(1/Vo)と基質濃度(1/[S])を逆数としてプロットします。このプロットにより、VmaxとKmを正確に評価できる直線が形成されます。
- VmaxとKmの正確な決定に役立ちます。
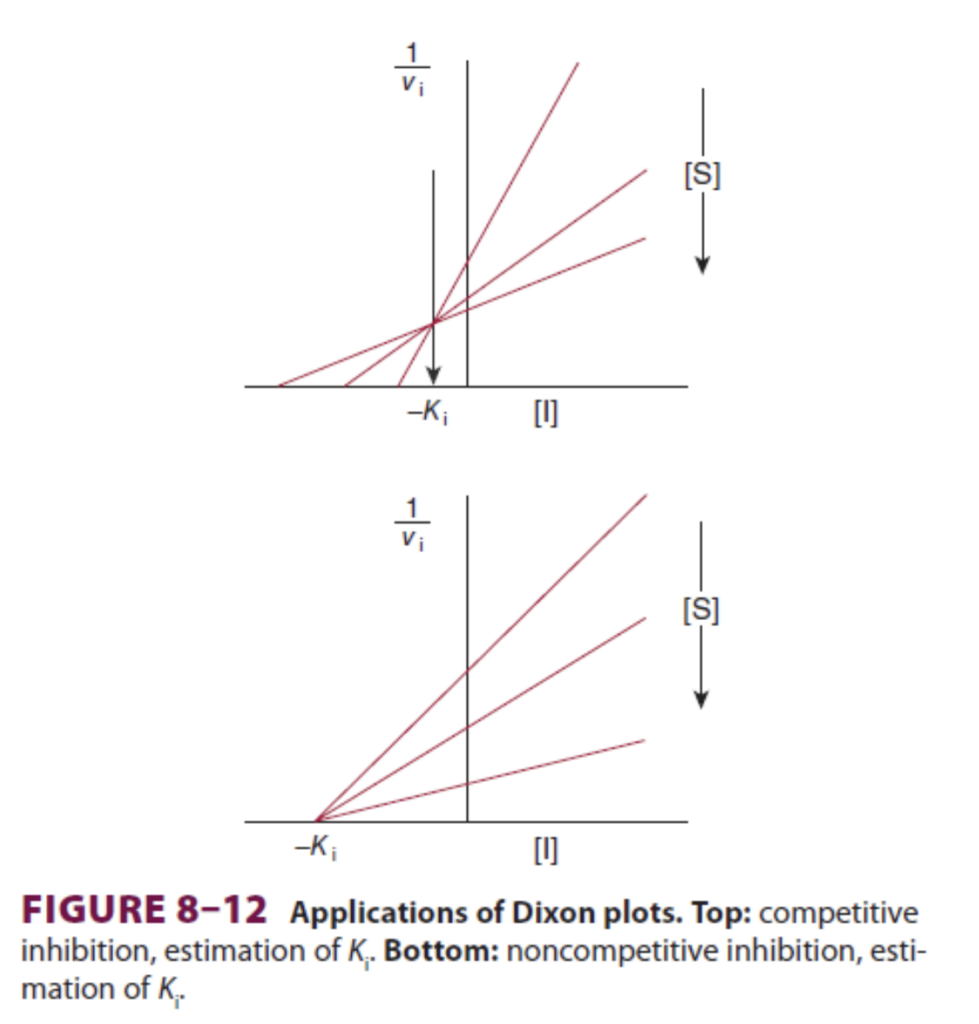
Dixonプロット
- Dixonプロットは、Lineweaver-Burkeプロットの代替手段です。こちらは基質濃度ではなく**阻害剤濃度(Inhibitor Concentration)**を使用してプロットします。
酵素阻害の種類(Types of Enzyme Inhibition)
酵素阻害には2つの大きな種類があります:
- 可逆的阻害(Reversible Inhibition)
- 競争的阻害(Competitive Inhibition)
- 非競争的阻害(Noncompetitive Inhibition)
- 反競争的阻害(Uncompetitive Inhibition)
- 不可逆的阻害(Irreversible Inhibition)
- 共有結合による酵素修飾(Covalent Enzyme Modification)
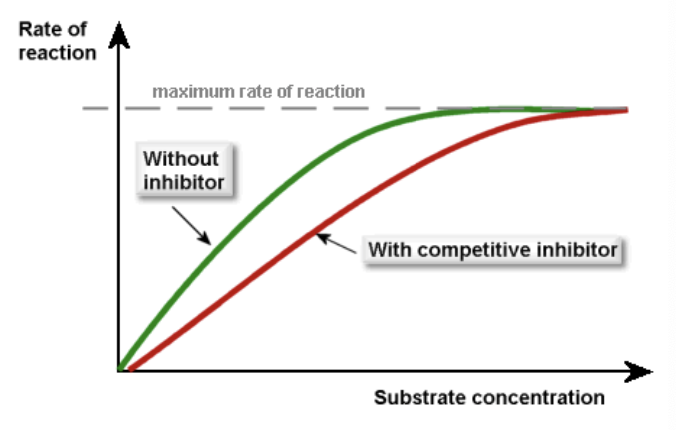
競争的阻害(Competitive Inhibition)
- **競争的阻害剤(Competitive Inhibitor)は、基質と活性部位(Active Site)**で競合します。阻害剤が活性部位に結合することで、基質が結合できなくなります。
- この結合は可逆的であり、基質濃度を増加させることで阻害剤の影響を打ち消すことができます。
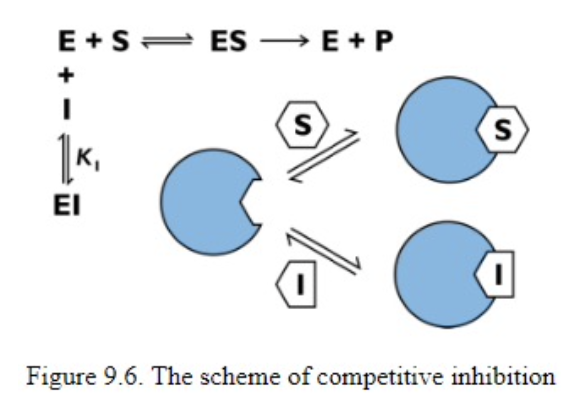
競争的阻害におけるKmとVmaxへの影響
- Km(ミカエリス定数): 増加します。基質が活性部位に結合するためには、より高い濃度が必要になります。
- Vmax(最大反応速度): 変化しません。十分な基質濃度を加えれば、最大速度に達します。
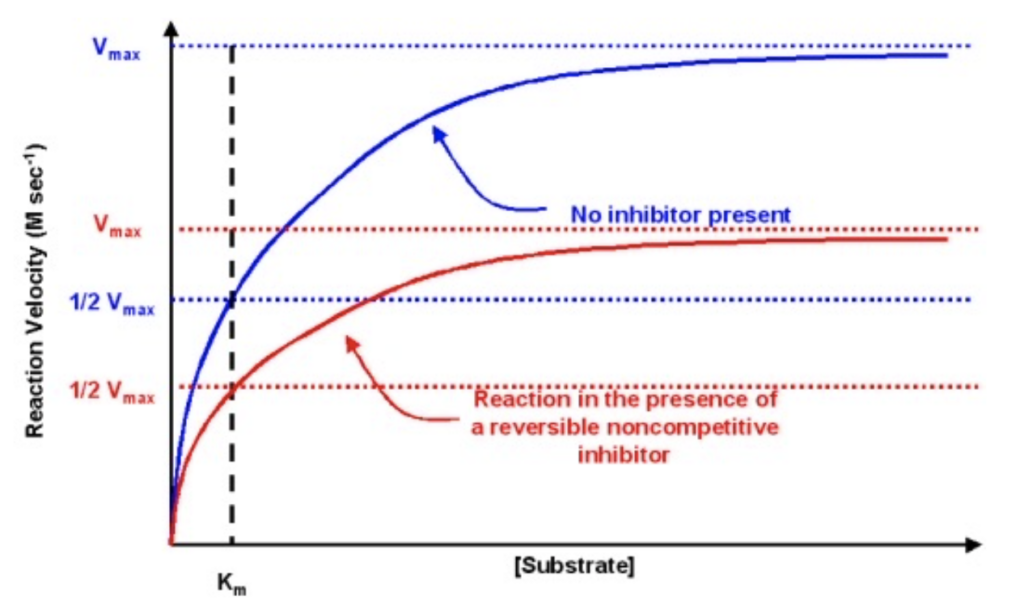
非競争的阻害(Noncompetitive Inhibition)/混合型阻害(Mixed Inhibition)
- **非競争的阻害剤(Noncompetitive Inhibitor)は、酵素のアロステリック部位(Allosteric Site)**に結合し、活性部位以外の場所で酵素の活性を妨げます。基質濃度が増加しても、この阻害を回避することはできません。
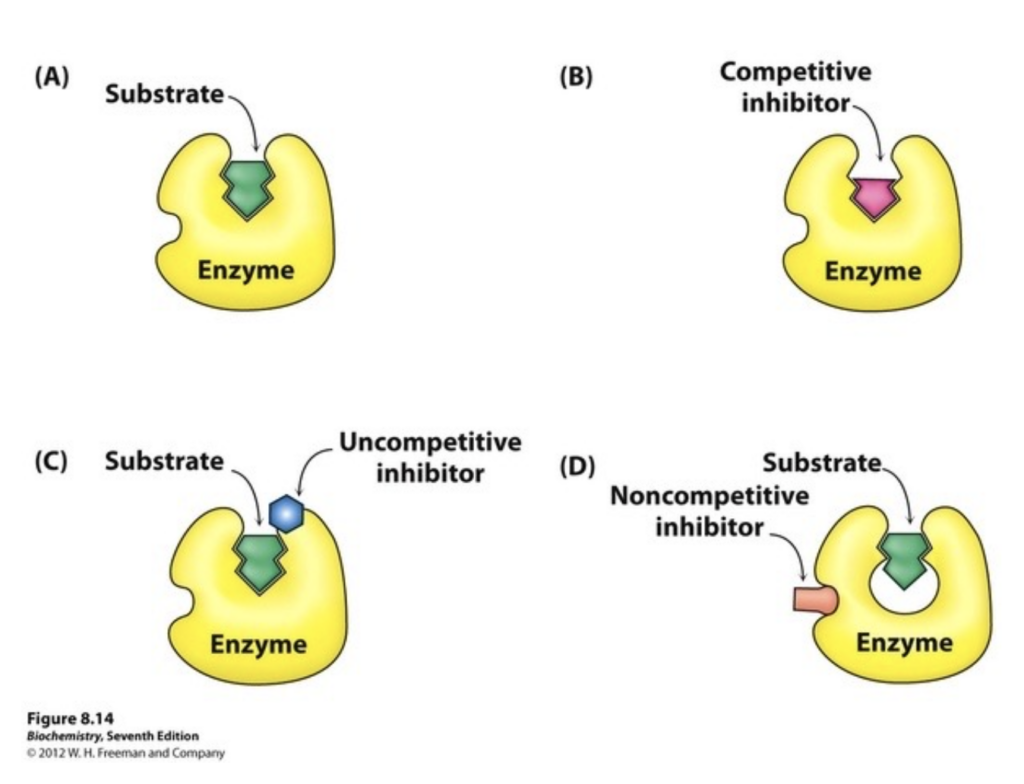
非競争的阻害におけるKmとVmaxへの影響
- Km: 変化しません。基質の結合は阻害されないため、Kmは一定です。
- Vmax: 減少します。全体的な反応速度が減少するため、最大速度も低下します。
反競争的阻害(Uncompetitive Inhibition)
- **反競争的阻害剤(Uncompetitive Inhibitor)**は、酵素-基質複合体(ES Complex)にのみ結合し、反応を妨げます。
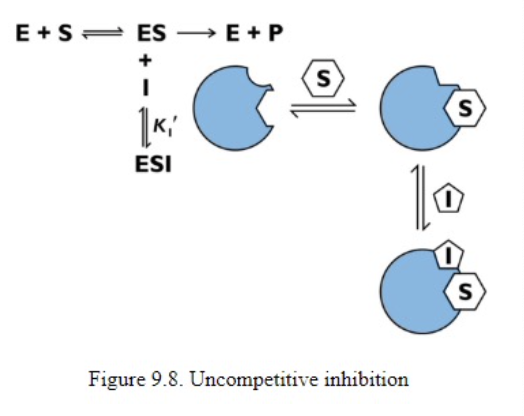
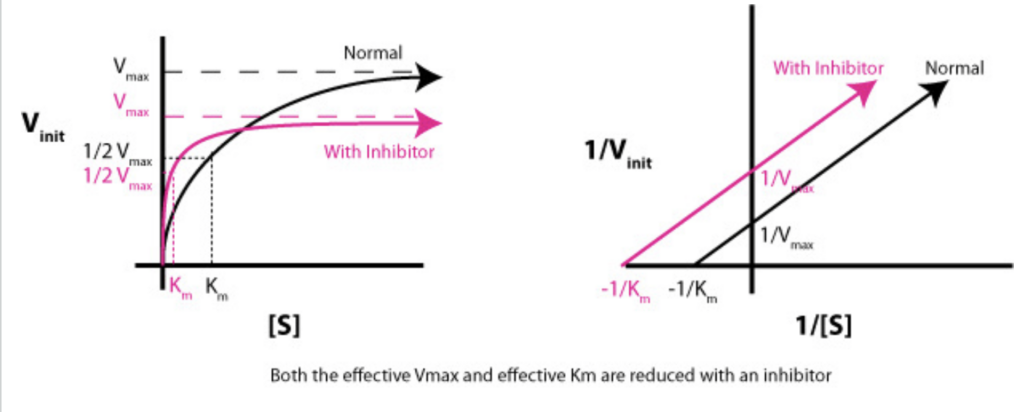
反競争的阻害におけるKmとVmaxへの影響
- Km: 減少します。酵素-基質複合体が安定化するため、基質の親和性が高くなり、Kmが減少します。
- Vmax: 減少します。反応速度自体が遅くなるため、Vmaxも低下します。
阻害タイプごとのKmとVmaxへの影響
| 阻害タイプ | Km | Vmax |
|---|---|---|
| 競争的阻害 | 増加 | 変わらない |
| 非競争的阻害 | 変わらない | 減少 |
| 反競争的阻害 | 減少 | 減少 |
競争的阻害(Competitive Inhibition)
- Km(ミカエリス定数): 増加
競争的阻害では、基質(Substrate)と阻害剤が同じ活性部位を競って結合します。阻害剤が結合していると基質が結合しにくいため、反応速度を上げるためにはより高い基質濃度が必要になり、結果としてKmが増加します。 - Vmax(最大反応速度): 変わらない
競争的阻害は高い基質濃度で打ち消すことができるため、十分な基質があれば酵素は最大速度に達することができ、Vmaxには影響がありません。
非競争的阻害(Noncompetitive Inhibition)
- Km: 変わらない
非競争的阻害では、阻害剤が基質とは**別の部位(アロステリック部位)**に結合するため、基質の結合自体には影響しません。したがって、Kmは変わりません。 - Vmax: 減少
阻害剤が結合すると、酵素の活性が低下して反応が遅くなります。そのため、どれだけ基質濃度を増やしても最大速度には達せず、Vmaxは減少します。
反競争的阻害(Uncompetitive Inhibition)
- Km: 減少
反競争的阻害では、阻害剤は**酵素-基質複合体(ES複合体)**に結合します。これにより、基質が酵素に結合している間、阻害剤がES複合体を安定化させるため、Kmが減少します(酵素が基質に対してより高い親和性を示すようになります)。 - Vmax: 減少
反競争的阻害では、酵素-基質複合体に阻害剤が結合するため、最大速度に達することが難しくなり、結果としてVmaxは減少します。
なぜ酵素活性を調節するのか?(Why Regulate Enzyme Activity?)
- 酵素活性の調節は、酵素の正と負の影響をコントロールし、**代謝効率(Metabolic Efficiency)**を最適化するために行われます。
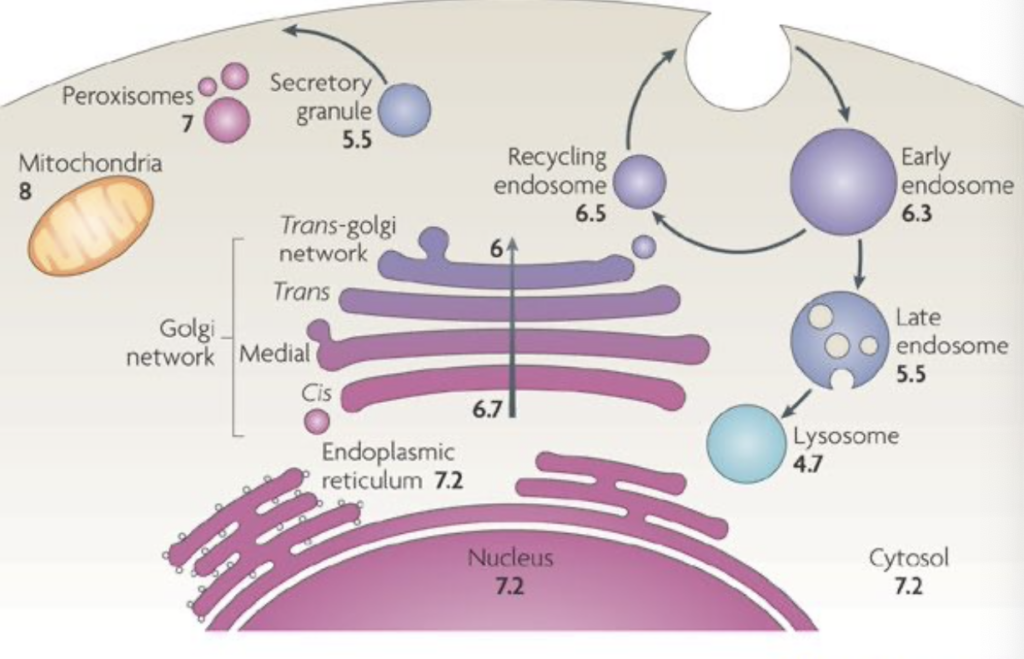
- コンパートメンタリゼーション
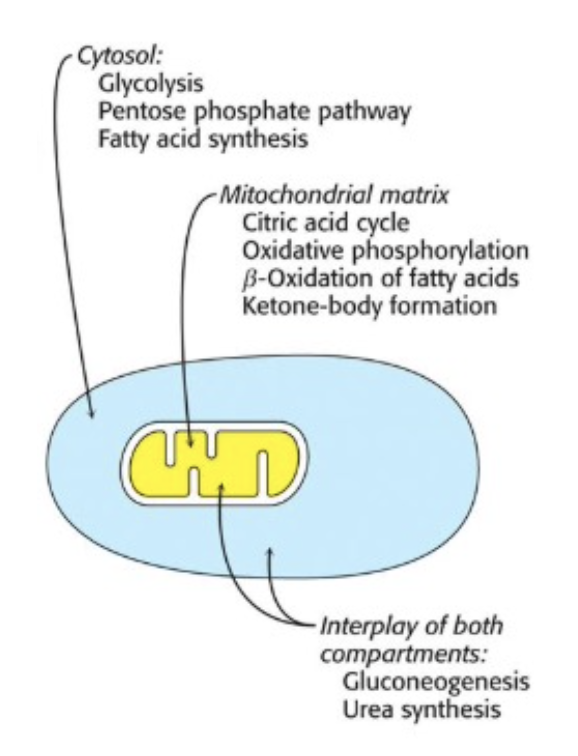
コンパートメンタリゼーション(Compartmentalization)
- コンパートメンタリゼーションは、酵素反応を特定の細胞区画やオルガネラに限定することを意味します。これにより、代謝が効率的に行われ、調節が簡単になります。
コンパートメンタリゼーションの例
- 多くの酵素は**リソソーム(Lysosome)**内に存在し、タンパク質や多糖類の分解に関与します。
- **脂肪酸合成(Fatty Acid Synthesis)**は細胞質(Cytosol)で行われ、一方で脂肪酸分解(Fatty Acid Oxidation)はミトコンドリア内で行われます。
コンパートメンタリゼーションの利点
- 酵素反応は、特定の区画内で効率的に行われるため、全体的な細胞機能が最適化されます。
- リソソームなどのオルガネラは、自己消化を防ぐためにリチック酵素の活動を制御します。
- 特定の基質の出入りを制御することで、酵素反応を調節できます。
コンパートメンタリゼーションの欠点
- エネルギー消費の増加。ATP依存のトランスポーターを頻繁に使用する必要があり、エネルギーの需要が増加します。
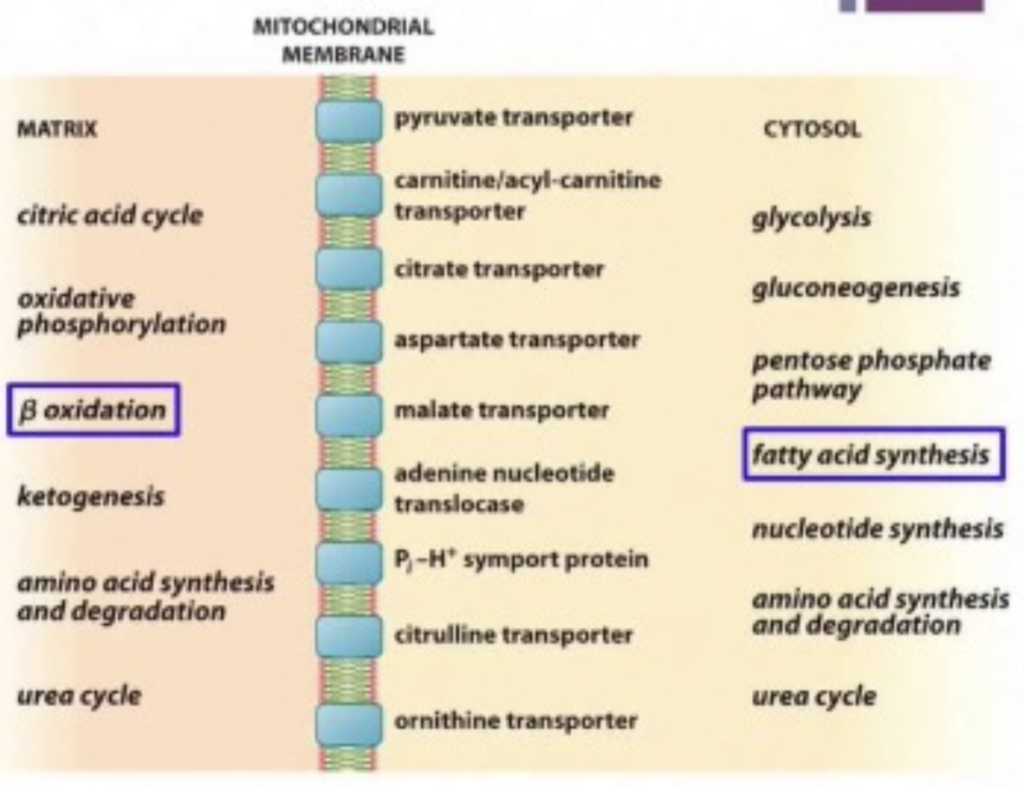
MECHANISMS FOR ENZYME REGULATION
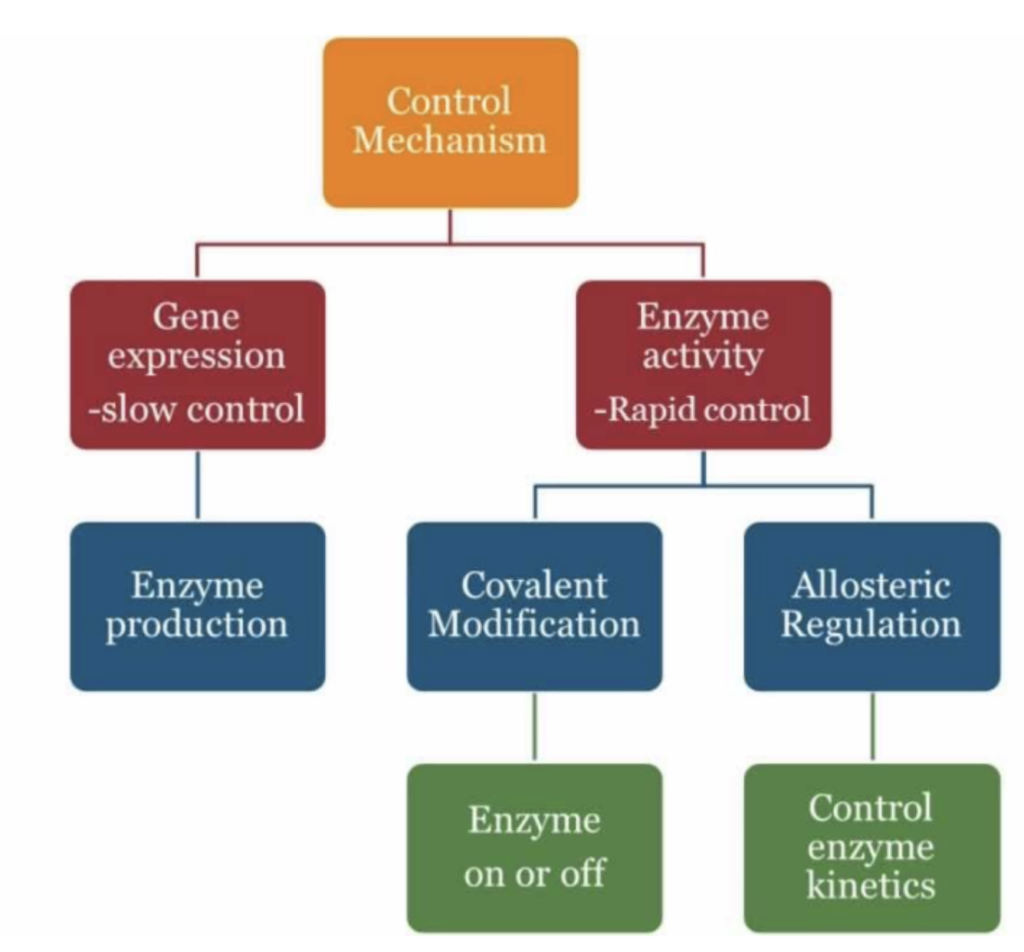
この図は、**酵素の制御メカニズム(Control Mechanism)**を示しています。酵素の制御は大きく2つの方法で行われ、**遅い制御(slow control)と迅速な制御(rapid control)**に分類されます。
1. 遅い制御(Slow Control): 遺伝子発現(Gene Expression)
- **遺伝子発現(Gene Expression)を介した酵素制御は、酵素の生成(Enzyme Production)**を制御します。これは遅い制御プロセスであり、酵素の合成が必要な状況で徐々に調整されます。たとえば、特定の条件下で必要な酵素が転写や翻訳のプロセスを経て産生されることが該当します。
2. 迅速な制御(Rapid Control): 酵素活性(Enzyme Activity)
- 酵素の活性化や不活性化を介して素早く反応を制御する方法です。これは、既存の酵素に対して迅速に反応を調整する必要がある場合に使われます。この迅速な制御には、2つの主要なメカニズムがあります。
- 共有結合修飾(Covalent Modification):
- 酵素の共有結合修飾により、酵素がオンまたはオフになります。例えば、リン酸化やアセチル化などの化学修飾によって酵素の活性が変わります。
- アロステリック調節(Allosteric Regulation):
- **アロステリック部位(Allosteric Site)に他の分子が結合することで、酵素の速度論的特性(Kinetics)**が制御されます。アロステリック調節により、酵素の活性を抑制または促進することができます。
- 共有結合修飾(Covalent Modification):
酵素の調節メカニズム(Mechanisms for Enzyme Regulation)
酵素の調節は大きく分けて、**遅い制御(Slow Control)と迅速な制御(Rapid Control)**の2つがあります。
遅い制御(Slow Control)
- 応答時間: 数分から数時間かかる
- この制御は、**遺伝子発現(Gene Expression)**を調整することで酵素の量を変化させるメカニズムです。たとえば、酵素の生成が減少し、分解が増加することにより酵素の量が減少します。
メカニズム
- 転写(Transcription): 遺伝子がmRNAに転写される過程
- 翻訳(Translation): mRNAがタンパク質に翻訳される過程
- 酵素ターンオーバー(Enzyme Turnover): 酵素が合成され、分解される速度の調整
遅い制御の具体例
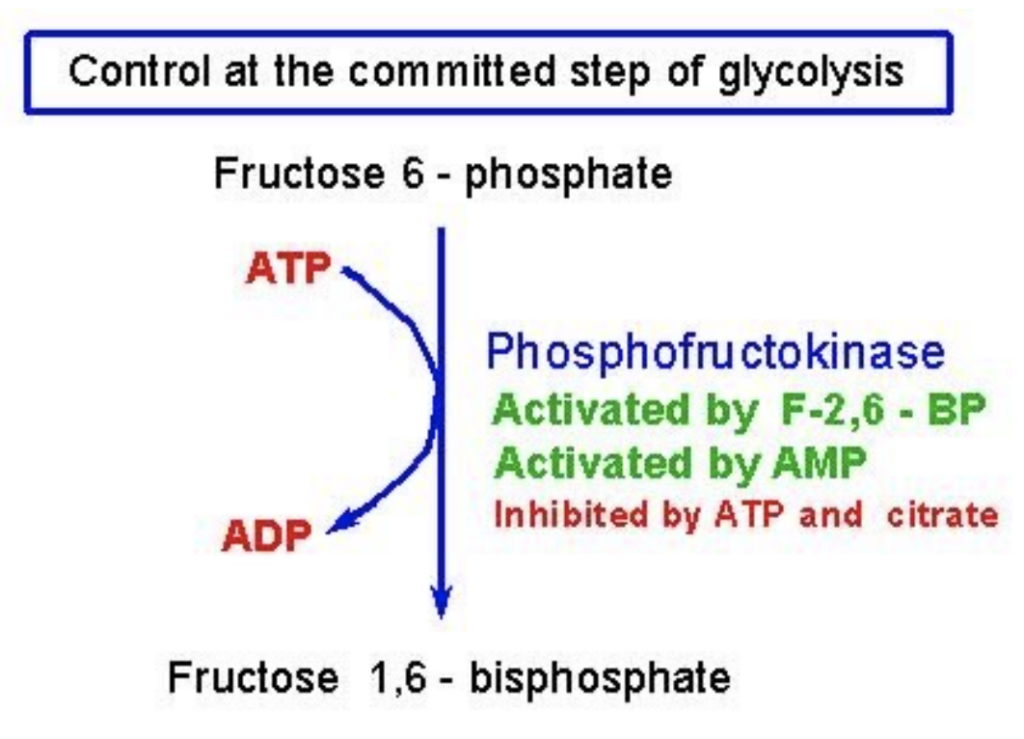
- 律速酵素の制御(Control of the Rate-Limiting Enzyme):
- **ホスホフルクトキナーゼ(Phosphofructokinase)**は、解糖系において不可逆な反応を触媒する律速酵素です。この酵素の調節が代謝の論理を決定します。
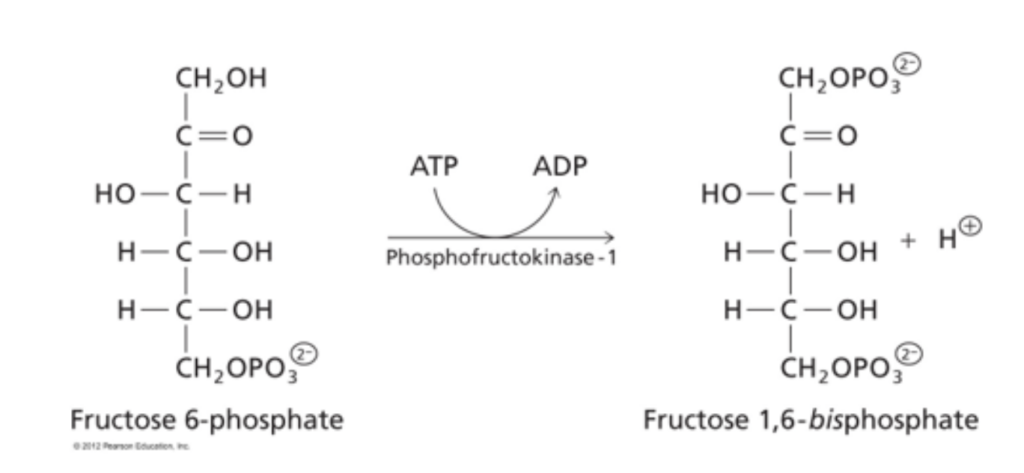
- 酵素合成の制御(Control of Enzyme Synthesis):
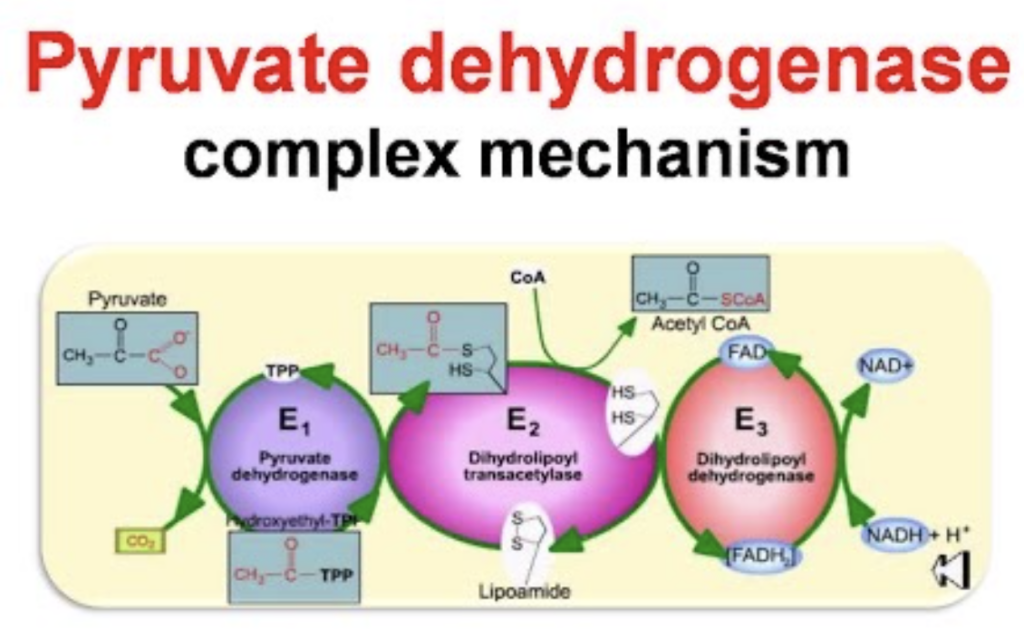
- 酵素の合成は、基質、補因子(Cofactor)、補酵素(Coenzyme)などのインデューサー(Inducer)の存在によって刺激される転写に依存しています。
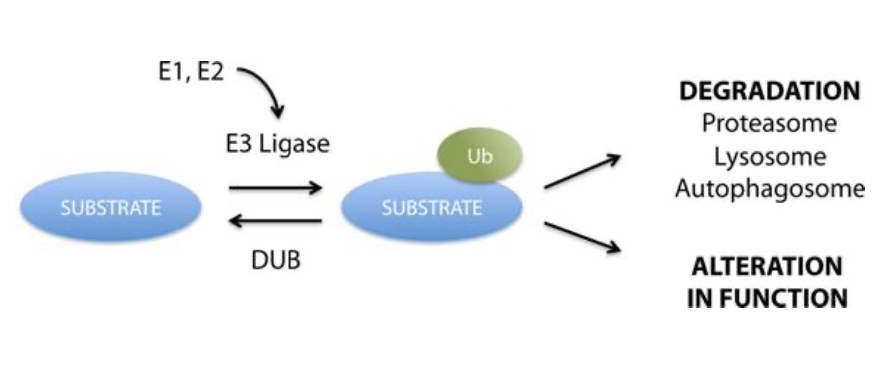
- 酵素分解の制御(Control of Enzyme Degradation):
- **ユビキチン化(Ubiquitination)**は、標的タンパク質分子にユビキチンが共有結合で付加される過程で、これにより標的タンパク質が分解されるマーカーとなります。
酵素合成の制御(Control of Enzyme Synthesis)
- 酵素の合成は、**インデューサー(Inducer)の存在に依存します。インデューサーは、基質(Substrate)、補因子(Cofactor)、補酵素(Coenzyme)などを指し、これらが遺伝子の転写(Transcription)**を刺激します。その結果、酵素が合成されます。
- 基質: 酵素が作用する分子
- 補因子: 酵素の機能に必要な無機分子やイオン
- 補酵素: 酵素の働きを助ける有機分子
このプロセスにより、特定の代謝経路や反応が必要なときに酵素の合成が促進されます。
酵素分解の制御(Control of Enzyme Degradation)
- **ユビキチン化(Ubiquitination)**は、タンパク質分解のための重要なメカニズムです。このプロセスでは、**ユビキチン分子(Ubiquitin Molecule)**がターゲットとなるタンパク質に共有結合で付加されます。ユビキチン化は、ターゲットタンパク質に「分解のマーカー(Marker for Protein Degradation)」を付ける役割を果たし、その後、**プロテアソーム(Proteasome)**で分解されます。
このプロセスは、不要なまたは損傷した酵素やタンパク質を細胞内から除去し、細胞の機能を維持するために重要です。
迅速な制御(Rapid Control)
- 応答時間: 数秒以内に起こる
- 酵素の活性を迅速に変化させるためのメカニズムで、酵素の量ではなく、既存の酵素の活性を調整します。
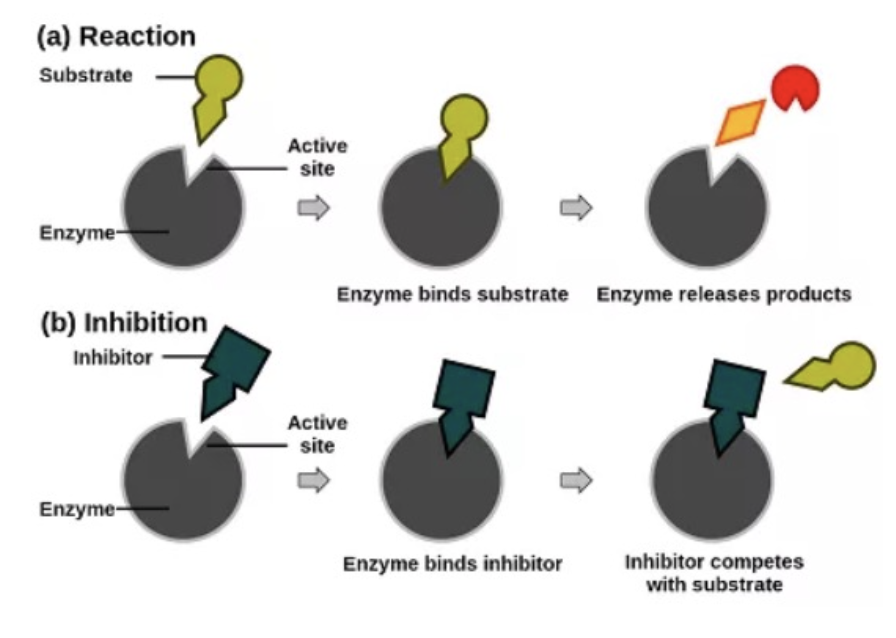
メカニズム
- アロステリック調節(Allosteric Regulation)
- 共有結合修飾(Covalent Modification)
- プロテオリティック活性化(Proteolytic Activation)
- フィードバック調節(Feedback Regulation)
アロステリック調節(Allosteric Regulation)
- アロステリック調節とは、**活性部位以外(アロステリック部位, Allosteric Site)**にエフェクター(Effector)やモジュレーター(Modulator)分子が結合することで酵素の活性が変化する仕組みです。
- アロステリック調節は、エフェクター分子の結合によって酵素の構造が変化し、酵素の活性が上昇または抑制されます。
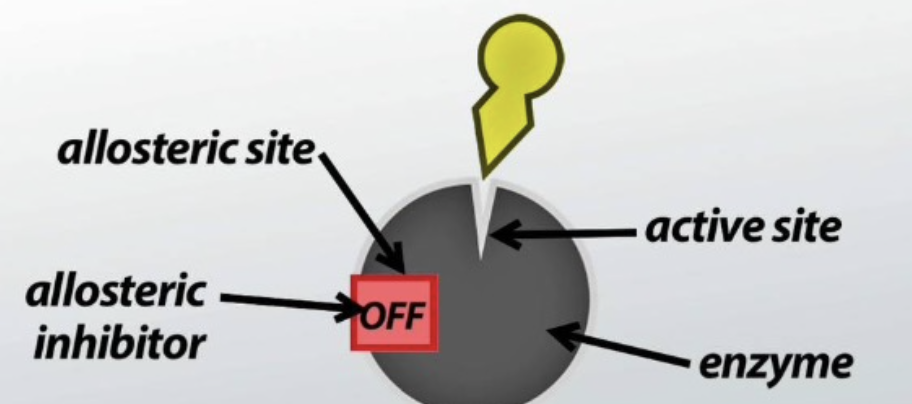
エフェクターの種類
- 正のエフェクター(Positive Effector): 酵素活性を増加させる
- 負のエフェクター(Negative Effector): 酵素活性を抑制する
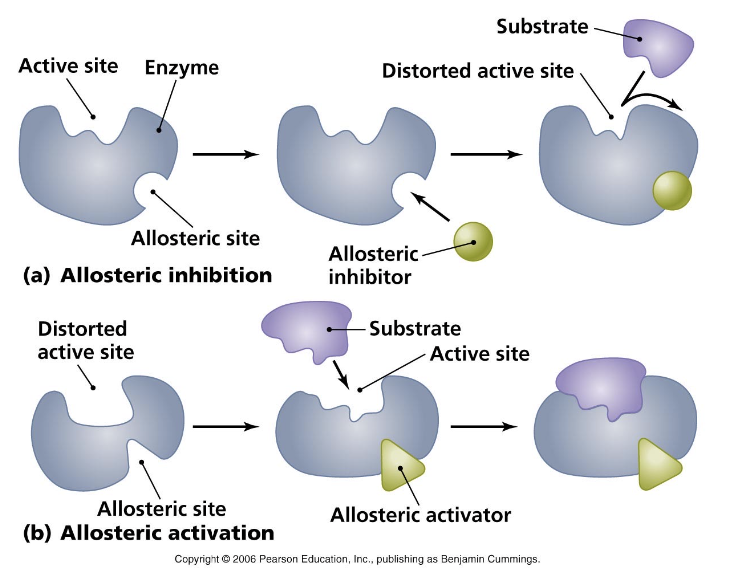
アロステリック調節の種類
- ホモトロピック調節(Homotropic Allosteric Modulation):
- 基質分子自体が調節を行う。多量体酵素(Multisubunit Enzymes)においてのみ起こり、**協同性結合(Cooperative Binding)**に従う。
- ヘテロトロピック調節(Heterotropic Allosteric Modulation):
- 基質以外の分子が調節を行う。
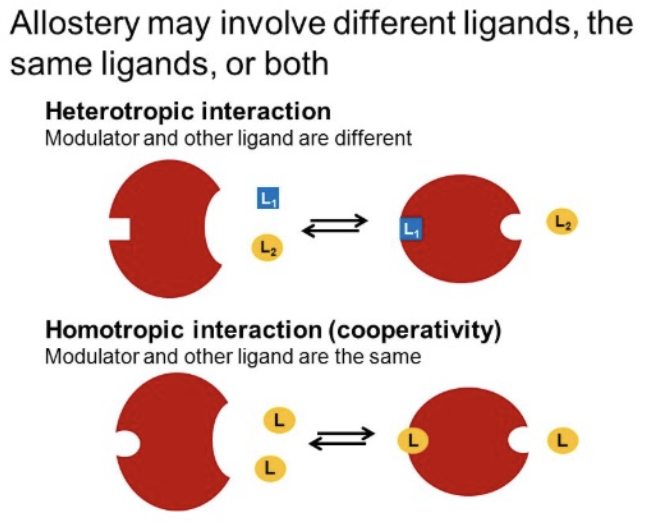
Hill式と協同結合(Hill Equation and Cooperative Binding)
- **Hill式(Hill Equation)**は、**協同結合(Cooperative Binding)**を記述する方程式です。これは、ヘモグロビンが酸素を結合する様子に似ており、特に多量体酵素(Multimeric Enzymes)が基質を複数の活性部位に結合する場合に見られる現象です。
- 協同結合は、1つの活性部位に基質が結合すると、他の活性部位への基質結合の親和性が変化するという性質を持っています。典型的な例として、**アスパラギン酸トランスカルバモイラーゼ(Aspartate Transcarbamoylase)**が挙げられます。
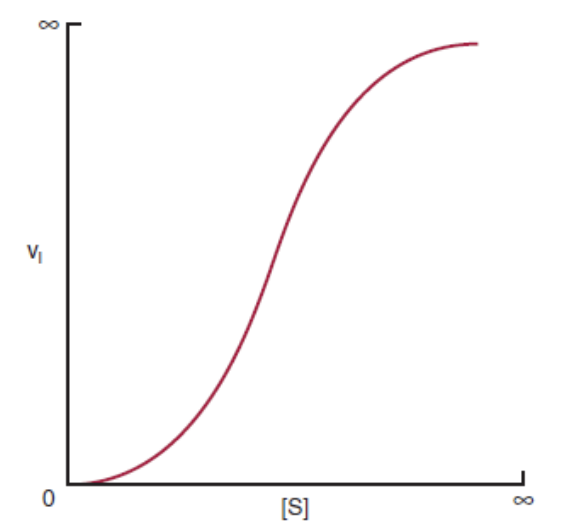
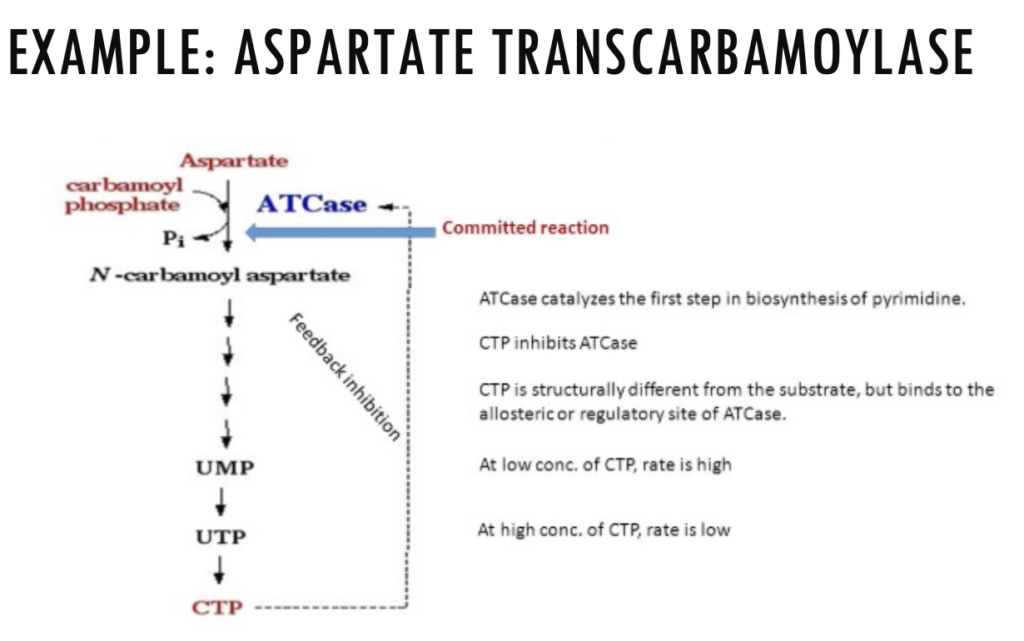
アロステリック調節とシグモイド曲線(Allosteric Regulation and Sigmoidal Curve)
- **アロステリック調節(Allosteric Regulation)**は、シグモイド曲線(Sigmoidal Curve)に従います。これは、基質濃度の増加に伴って、反応速度が急激に上昇する協同結合を示すものです。
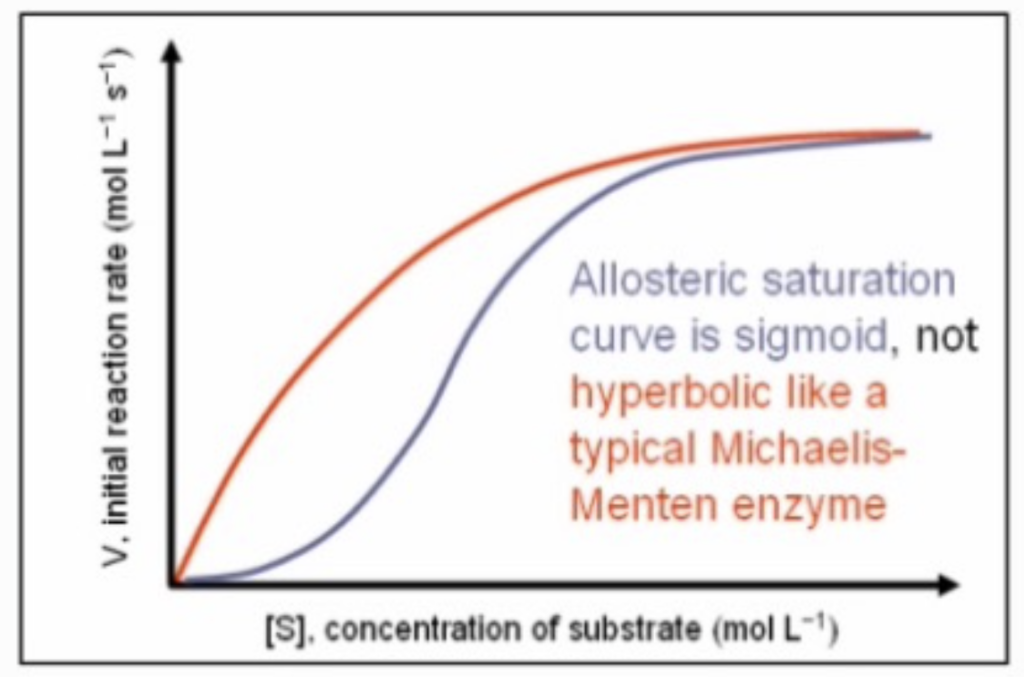
アロステリック調節によるKmとVmaxへの影響
- Km(ミカエリス定数): 増加することがあります。協同性が増すことで、より高い基質濃度が必要になる場合があります。
- Vmax(最大反応速度): 変わらないことが多いですが、調節の種類によっては変化することもあります。
共有結合修飾(Covalent Modification)
- 共有結合修飾は、可逆的または不可逆的に行われることがあります。酵素活性を迅速に調節するために使用されます。
共有結合修飾の例
- アセチル化(Acetylation)
- メチル化(Methylation)
- ADPリボシル化(ADP-Ribosylation)
- リン酸化(Phosphorylation)
プロテオリティック活性化(Proteolytic Activation)
- 一部のタンパク質は、**不活性前駆体タンパク質(プロタンパク質, Proproteins)**として合成されます。これらは、特定の条件下で活性化され、活性酵素になります。これにより、細胞が自己消化から保護されます。
プロ酵素(Zymogen)の例
- ペプシノーゲン(Pepsinogen) → ペプシン(Pepsin)
- トリプシノーゲン(Trypsinogen) → トリプシン(Trypsin)
- キモトリプシノーゲン(Chymotrypsinogen) → キモトリプシン(Chymotrypsin)
以下に、プロ酵素(Zymogen)、活性化因子(Activator)、および活性化された酵素(Active Enzyme)を表でまとめて解説します。
| プロ酵素(Zymogen) | 活性化因子(Activator) | 活性酵素(Active Enzyme) | 役割 |
|---|---|---|---|
| ペプシノーゲン(Pepsinogen) | H⁺、ペプシン(Pepsin) | ペプシン(Pepsin) | 胃でタンパク質を分解 |
| トリプシノーゲン(Trypsinogen) | トリプシン(Trypsin)、エンテロキナーゼ(Enterokinase) | トリプシン(Trypsin) | 小腸でタンパク質を分解、他のプロ酵素を活性化 |
| キモトリプシノーゲン(Chymotrypsinogen) | トリプシン(Trypsin) | キモトリプシン(Chymotrypsin) | 小腸でタンパク質を分解 |
| プロカルボキシペプチダーゼ(Procarboxypeptidase) | トリプシン(Trypsin) | カルボキシペプチダーゼ(Carboxypeptidase) | 小腸でペプチドのC末端アミノ酸を切断 |
| プロエラスターゼ(Proelastase) | トリプシン(Trypsin) | エラスターゼ(Elastase) | 結合組織中のエラスチンを分解 |
| プロレニン(Prorennin) | H⁺、レニン(Rennin) | レニン(Rennin) | 乳児の胃でミルクタンパク質(カゼイン)を凝固させる |
プロテオリティック活性化の重要なポイント
- プロ酵素のプロテオリティック活性化は、**生理的に不可逆的(Irreversible)**です。一度活性化された酵素は、その触媒活性を持続します。
フィードバック調節(Feedback Regulation)
- フィードバック調節は、経路の最終産物がその経路の初期段階に関与する酵素を阻害するプロセスです。これにより、経路全体の活動が調整されます。
- 例: 生成物Dの濃度が高い場合、酵素1(Enz1)によるAからBへの変換を阻害します。
例: ホスホフルクトキナーゼ(Phosphofructokinase)
- ホスホフルクトキナーゼは、解糖系の律速段階を調節し、フィードバック調節を受けます。
今日の学習目標(Learning Objectives for Today)
- Michaelis-Menten式とHill式を説明し、その用途を理解する
- 競争的阻害、非競争的阻害、反競争的阻害のメカニズムを理解し説明する
- Lineweaver-Burkeプロットを説明し、その用途を理解する
- 多くの酵素における細胞内基質濃度がKmに近い理由を説明する
- 酵素調節のさまざまな方法を列挙し、それぞれを説明する

コメント