
Contents
- 1 授業PPTより
- 1.1 病理学 (Pathology) の定義
- 1.2 全身病理学と系統病理学 (General Pathology and Systemic Pathology)
- 1.3 細胞のストレスに対する応答 (Cellular Response to Stress)
- 1.4 細胞の適応の種類 (Adaptive Changes Include)
- 1.5 細胞損傷の原因 (Causes of Cell Injury)
- 1.6 可逆的損傷 (Reversible Injury)
- 1.7 脂肪変性 (Fatty Change)
- 1.8 細胞死 (Cell Death)
- 1.9 壊死 (Necrosis)
- 1.10 壊死の組織パターン (Patterns of Tissue Necrosis)
- 1.11 アポトーシス (Apoptosis)
- 1.12 異常なカルシウム沈着 (Pathologic Calcification)
- 1.13 Giant Cell(多核巨細胞)
- 1.14 Granulomaにおける役割
- 2 細胞傷害、細胞死、適応
- 2.1 病理学の導入 (INTRODUCTION TO PATHOLOGY)
- 2.2 ストレスと有害刺激に対する細胞の反応の概要 (OVERVIEW OF CELLULAR RESPONSES TO STRESS AND NOXIOUS STIMULI)
- 2.3 細胞傷害の原因 (CAUSES OF CELL INJURY)
- 2.4 細胞傷害および細胞死の過程 (SEQUENCE OF EVENTS IN CELL INJURY AND CELL DEATH)
- 2.5 可逆的な細胞傷害 (Reversible Cell Injury)
- 2.6 形態学的変化 (MORPHOLOGY)
- 2.7 細胞死 (Cell Death)
- 2.8 壊死 (Necrosis)
- 2.9 形態学的変化 (MORPHOLOGY)
- 2.10 細胞膜の損傷によるタンパク質の漏出 (Leakage of intracellular proteins)
- 2.11 アポトーシス (Apoptosis)
- 2.12 アポトーシスの原因 (Causes of Apoptosis)
- 2.13 アポトーシスのメカニズム (Mechanisms of Apoptosis)
- 2.14 アポトーシス細胞の除去 (Clearance of apoptotic cells)
- 2.15 形態学的変化 (MORPHOLOGY)
- 2.16 その他の細胞死の経路 (Other Pathways of Cell Death)
- 2.17 オートファジー (Autophagy)
- 2.18 要約 (SUMMARY)
- 2.19 細胞傷害および細胞死のパターン (Patterns of Cell Injury and Cell Death)
- 2.20 細胞傷害および細胞死のメカニズム (Mechanisms of Cell Injury and Death)
- 2.21 低酸素症と虚血 (Hypoxia and Ischemia)
- 2.22 虚血再灌流傷害 (Ischemia-Reperfusion Injury)
- 2.23 毒素による細胞傷害 (Cell Injury Caused by Toxins)
- 2.24 小胞体ストレス (Endoplasmic Reticulum Stress)
- 2.25 DNA損傷 (DNA Damage)
- 2.26 炎症 (Inflammation)
- 2.27 多様な原因による細胞傷害の共通のイベント (Common Events in Cell Injury From Diverse Causes)
- 2.28 ミトコンドリア機能障害 (Mitochondrial Dysfunction)
- 2.29 膜透過性の欠陥 (Defects in Membrane Permeability)
- 2.30 要約 (SUMMARY)
- 2.31 細胞傷害のメカニズム (Mechanisms of Cell Injury)
- 2.32 ストレスに対する細胞の適応 (Cellular Adaptations to Stress)
- 2.33 肥大 (Hypertrophy)
- 2.34 過形成 (Hyperplasia)
- 2.35 萎縮 (Atrophy)
- 2.36 化生 (Metaplasia)
- 2.37 ストレスに対する細胞の適応 (Summary: Cellular Adaptations to Stress)
- 2.38 細胞内蓄積物 (Intracellular Accumulations)
- 2.39 病理学的石灰化 (Pathologic Calcification)
- 2.40 形態学 (Morphology)
- 2.41 要約 (Summary)
- 2.42 異常な細胞内沈着物および石灰化 (Abnormal Intracellular Depositions and Calcifications)
- 2.43 細胞の老化 (Cellular Aging)
- 2.44 要約 (Summary)
- 2.45 細胞の老化 (Cellular Aging)
授業PPTより
病理学 (Pathology) の定義
- 病理学では、細胞、組織、臓器の構造的、化学的、生理的な変化について研究し、これらがどのように疾患の基礎となっているかを理解します(structural, biochemical, and functional changes)。
全身病理学と系統病理学 (General Pathology and Systemic Pathology)
病気には4つの主要な側面があります。
- 原因 (Etiology): 遺伝的要因または環境的要因(genetic or environmental)。
- 発症機序 (Pathogenesis): 生化学的および分子レベルでの発病メカニズム(biochemical, molecular mechanisms)。
- 形態学的変化 (Morphologic Changes): 細胞や臓器における構造的変化(structural alterations)。
- 臨床的な結果 (Clinical Manifestations): これらの変化による影響(consequences of changes)。
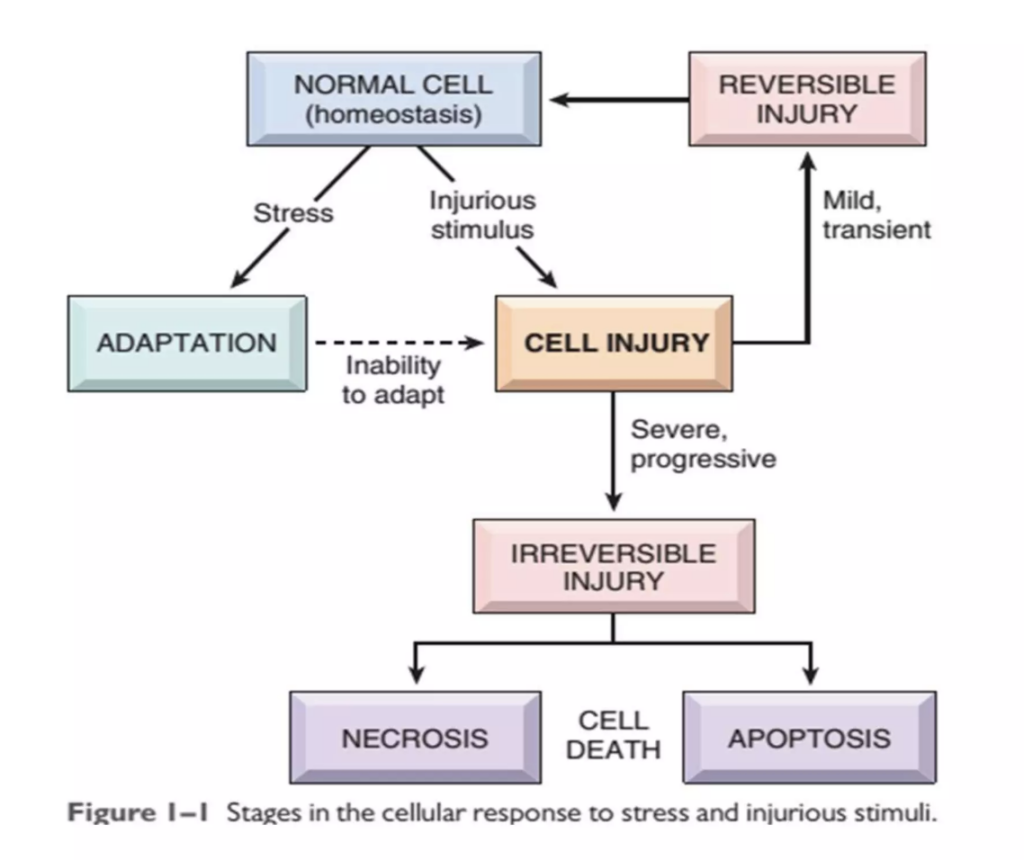
この図は、細胞がストレスや傷害性の刺激に対してどのように反応するかを示したフローチャートです。以下のように、正常な細胞が異なる段階を経て、最終的に回復するか、不可逆的な細胞死に至るかを説明しています。
- 正常な細胞 (Normal Cell): 細胞は通常、恒常性 (homeostasis) を保ちながら安定した状態にあります。
- ストレス (Stress): 細胞に対してストレスが加わると、細胞は 適応 (Adaptation) します。例えば、細胞の大きさが変わったり、機能を変化させてストレスに対応します。適応が成功すれば、細胞は引き続き生存します。
- 傷害性刺激 (Injurious Stimulus): ストレスが強すぎたり、長時間にわたると、適応が不十分で細胞は 損傷 (Cell Injury) を受けます。
- 可逆的損傷 (Reversible Injury): 細胞損傷が軽度で一時的であれば、細胞は回復し、再び正常な状態に戻ることができます。この段階では、損傷は完全に
修復可能であり、細胞の機能は回復します。
- 不可逆的損傷 (Irreversible Injury): 損傷が重度で進行してしまった場合、細胞はもはや修復できず、不可逆的な損傷となります。この段階では、細胞は死に向かいます。
- 細胞死 (Cell Death): 細胞死には2つの形態があります:
- ネクローシス (Necrosis): 病的な細胞死で、通常は炎症反応を伴い、周囲の組織にも影響を及ぼします。
- アポトーシス (Apoptosis): 細胞の自己制御による計画的な細胞死で、炎症を引き起こさずに細胞が整理されます。
細胞のストレスに対する応答 (Cellular Response to Stress)
- 適応 (Adaptation): 細胞が生理的または病的状態に対して行う可逆的な機能的および構造的な変化です。これにより、細胞は生き残り、機能を続けることができます(Reversible functional and structural changes)。
細胞の適応の種類 (Adaptive Changes Include)
- 肥大 (Hypertrophy): 細胞のサイズが増加する(Increase in size of cells)。
- 過形成 (Hyperplasia): 細胞の数が増加する(Increase in number of cells)。
- 萎縮 (Atrophy): 細胞のサイズが減少する(Decrease in size of cells)。
- 化生 (Metaplasia): 細胞の表現型が変化する。ある細胞のタイプが別の細胞タイプに置き換わる(Change in phenotype of cells)。
細胞損傷の原因 (Causes of Cell Injury)
- 酸素欠乏(Oxygen deprivation)
- 物理的要因(Physical agents: trauma, burns, temperature, radiation, electric shock)
- 化学物質と薬物(Chemicals and Drugs)
- 感染性因子(Infectious agents)
- 免疫反応(Immune reactions)
- 栄養不均衡(Nutritional imbalances)
適応の限界を超えると細胞損傷が発生し、損傷が軽度または一時的であれば可逆的ですが、重度または持続的な場合は不可逆的な損傷、さらには細胞死に至る可能性があります(Cell injury is reversible up to a certain point. If stimulus is severe or prolonged, irreversible injury occurs leading to cell death)。
可逆的損傷 (Reversible Injury)
光学顕微鏡で観察される可逆的な細胞損傷の特徴:
- 細胞の膨潤 (Cellular Swelling): 細胞内に水が入り込むことによる膨潤。初期には小さな空胞が見られ、細胞質は赤く見える(Cytoplasm appears red)。
- 脂肪変性 (Fatty Change): 脂質代謝に関与する臓器(肝臓など)で脂質の蓄積が見られる(Fatty Change in organs involved in lipid metabolism)。
脂肪変性 (Fatty Change)
- 脂肪肝 (Fatty Liver): 肝臓で中性脂肪を含む脂肪空胞が蓄積し、肝細胞が肥大して核が片側に押しやられることが特徴です(Hepatic steatosis)。
細胞死 (Cell Death)
細胞死には主に2つのタイプがあります。
- 壊死 (Necrosis): 無制御な細胞死で、炎症を引き起こす(Unregulated cell death, inflammation)。
- アポトーシス (Apoptosis): プログラム化された細胞死で、時には生理的プロセスとして起こる(Programmed cell death, physiological process)。
壊死 (Necrosis)
- 壊死は病理学的なプロセスであり、細胞が重度の損傷を受けた結果として起こります(Pathological process due to severe injury)。
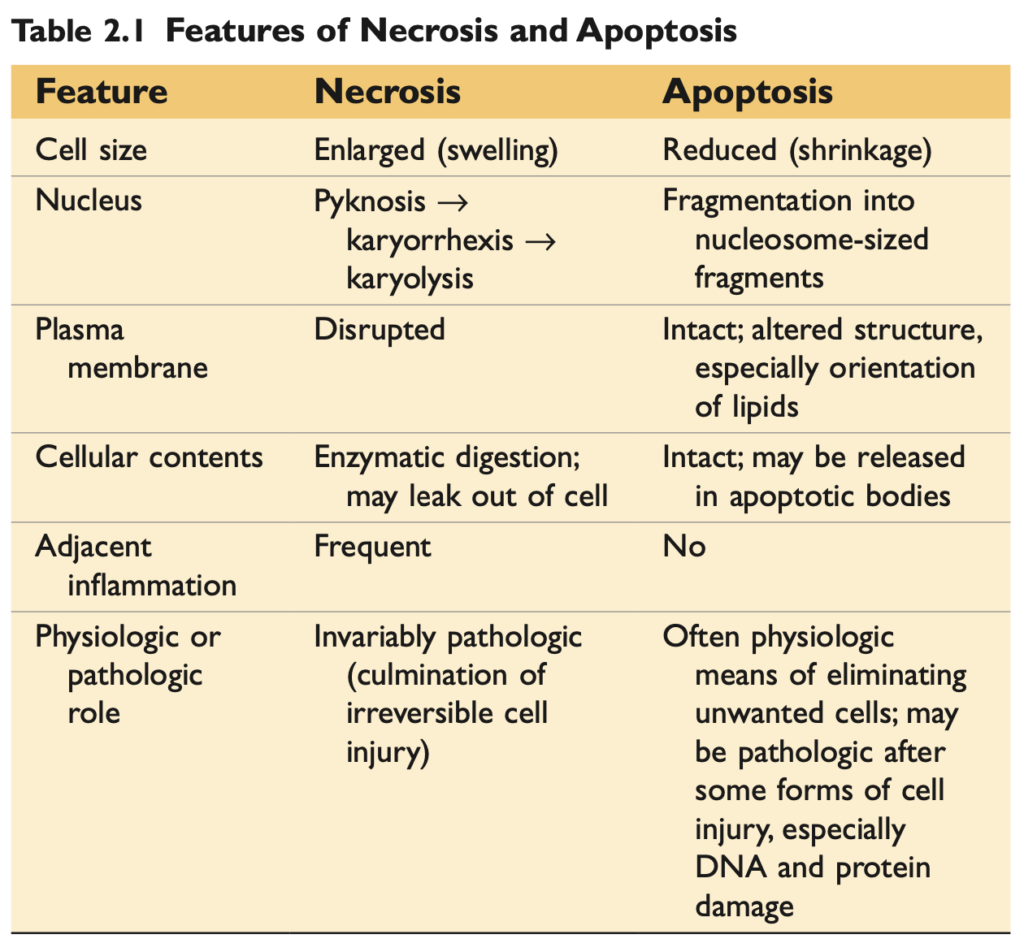
- 壊死した細胞は炎症を引き起こし、細胞内容物が漏れ出します。核の変化として融解核 (Karyolysis)、縮小核 (Pyknosis)、および**核破片化 (Karyorrhexis)**が見られます。
| Feature (特徴) | Necrosis (ネクローシス) | Apoptosis (アポトーシス) |
|---|---|---|
| Cell size (細胞の大きさ) | Enlarged (swelling) / 拡大(膨張) | Reduced (shrinkage) / 縮小(収縮) |
| Nucleus (核) | Pyknosis → karyorrhexis → karyolysis / 核濃縮 → 核断片化 → 核融解 | Fragmentation into nucleosome-sized fragments / ヌクレオソームサイズの断片へ分解 |
| Plasma membrane (細胞膜) | Disrupted / 破壊される | Intact; altered structure, especially lipid orientation / 保持されるが、構造が変化し、特に脂質の配置が変わる |
| Cellular contents (細胞内容物) | Enzymatic digestion; may leak out of cell / 酵素による分解、細胞外に漏れる場合がある | Intact; may be released in apoptotic bodies / 保持されるが、アポトーシス小体として放出される場合がある |
| Adjacent inflammation (隣接する炎症) | Frequent / よく見られる | No / 見られない |
| Physiologic or pathologic role (生理的・病理的役割) | Invariably pathologic (culmination of irreversible cell injury) / 常に病理的(不可逆的な細胞傷害の結果) | Often physiologic means of eliminating unwanted cells; may be pathologic after some forms of cell injury, especially DNA and protein damage / 不要な細胞を除去する生理的な手段であることが多いが、特にDNAやタンパク質損傷によって病理的になる場合もある |
壊死の組織パターン (Patterns of Tissue Necrosis)
壊死が大規模に起こると、臓器全体が壊死したとされます。壊死にはいくつかの異なるパターンがあります。
- 凝固壊死 (Coagulative Necrosis): 細胞の構造が保存され、組織は硬い質感を持つ(Firm texture)。
- 脂肪壊死 (Fat Necrosis): 通常、膵臓酵素の活性化により脂肪組織が破壊される(Fat destruction by pancreatic enzymes)。
- 融解壊死 (Liquefactive Necrosis): 細胞が液化し、粘稠な質感の物質が形成される(Viscous mass from liquefied dead cells)。
- 乾酪壊死 (Caseous Necrosis): 結核や真菌感染で見られる白色でチーズ様の外観(Cheese-like appearance in tuberculosis)。
アポトーシス (Apoptosis)
- アポトーシスでは細胞の縮小、クロマチン凝縮、アポトーシス小体の形成などの変化が見られます。炎症反応を伴わないことが特徴です(Cell shrinkage, chromatin condensation, apoptotic bodies without inflammation)。
異常なカルシウム沈着 (Pathologic Calcification)
異常な組織内でカルシウム塩が沈着する現象には2つのタイプがあります。
- 栄養障害性石灰化 (Dystrophic Calcification): 壊死した組織にカルシウムが沈着し、通常は血清カルシウム濃度は正常(Serum calcium is normal)。
- 転移性石灰化 (Metastatic Calcification): 血中カルシウム濃度が上昇した場合に正常組織にカルシウムが沈着する(Hypercalcemia leads to calcification in normal tissues)。
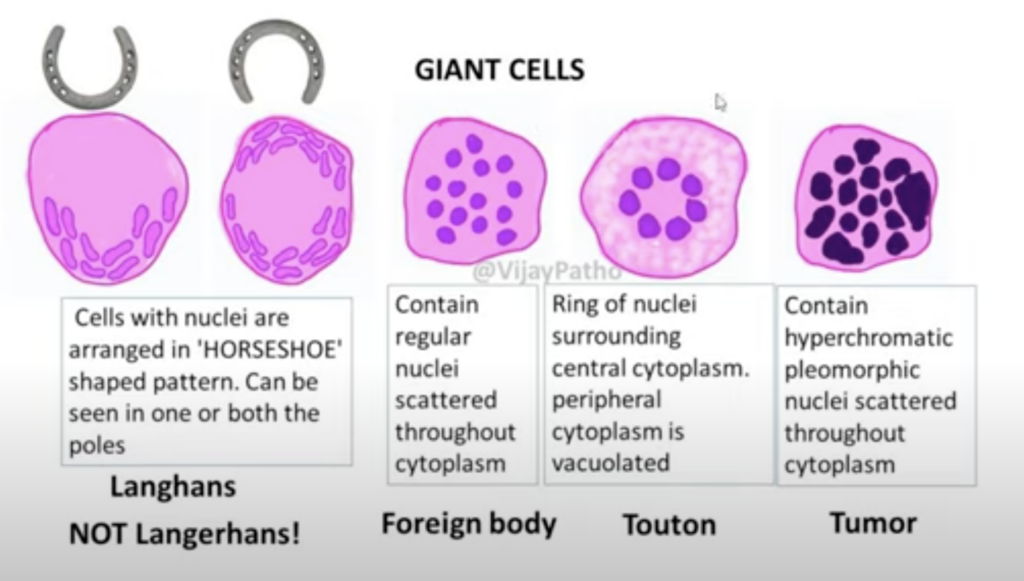
Giant Cell(多核巨細胞)
多核巨細胞とは、複数のマクロファージが融合してできた大きな細胞で、1つの細胞内に多くの核を持つのが特徴です。これらは肉芽腫の中心部分に存在することが多く、異物や感染病原体を封じ込める役割を果たしています。
Giant Cellの役割
- 異物の排除: 巨細胞は異物を隔離・消化するために形成されますが、特に大きな異物や難治性の病原体に対して、マクロファージ単体では対処しきれないため、これらが融合して1つの大きな細胞として機能します。
- 慢性炎症反応: 長期間にわたる炎症刺激や特定の感染症に対する防御反応の一環としても見られます。特に、結核菌や真菌、シリカなどの持続的な刺激に対して出現します。
Giant Cellの種類
- Langhans Giant Cell(ラングハンス型巨細胞):
- 核が細胞の周縁部に馬蹄形や環状に配置されているのが特徴。
- 主に結核やサルコイドーシスなどの肉芽腫性疾患で見られます。
- Foreign Body Giant Cell(異物型巨細胞):
- 核が不規則に細胞全体に散在しています。
- 異物に反応して形成され、異物の近くに見られることが多いです。例えば、縫合糸やシリコン、鉱粉などの異物が体内に入った場合に出現します。
Granulomaにおける役割
Granulomaは、体が異物や難治性の病原体を完全に排除できない場合に、これらを隔離するために形成されます。多核巨細胞はその中心にあり、異物や病原体を封じ込めて局所に炎症を維持し、周囲の組織への拡大を防ぐ役割を担っています。
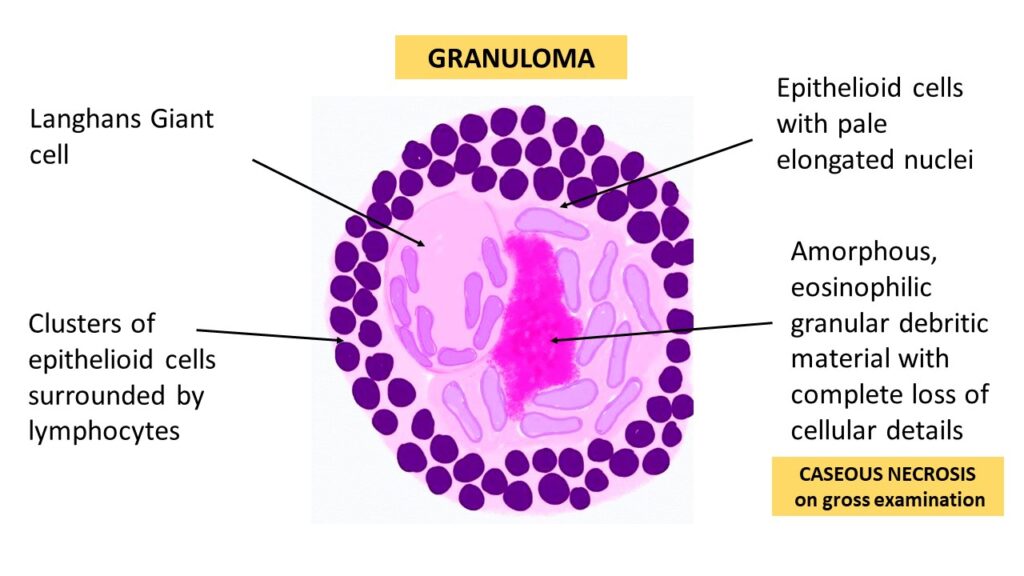
この図は**肉芽腫 (Granuloma)**の構造を示しています。肉芽腫は、異物や持続的な感染に対する体の慢性炎症反応として形成されます。図中に示されている重要な要素について解説します。
- Langhans Giant Cell(ラングハンス型巨細胞):
- 複数のマクロファージが融合して形成される多核巨細胞の一種です。この細胞では、核が馬蹄形または円形に周縁に集まっているのが特徴です。特に結核やサルコイドーシスなどの慢性肉芽腫性疾患で見られます。
- Clusters of epithelioid cells surrounded by lymphocytes(リンパ球に囲まれた上皮様細胞の集まり):
- 上皮様細胞 (Epithelioid cells) は、マクロファージが活性化し、形態が上皮細胞に似た細胞に変化したもので、肉芽腫の形成に関与します。これらの細胞は、肉芽腫の中心部に集まり、感染や異物に対する防御を行います。
- リンパ球 (Lymphocytes) は、免疫応答の重要な一部であり、周囲を囲んでいます。
- Epithelioid cells with pale elongated nuclei(淡い長い核を持つ上皮様細胞):
- 上皮様細胞は長くて淡い核を持つことがあり、これにより異物や持続的な刺激に対抗します。
- Amorphous, eosinophilic granular debritic material with complete loss of cellular details(無形の、好酸性の顆粒状の壊死物質、細胞の詳細が完全に失われている):
- これは**乾酪壊死 (Caseous Necrosis)**と呼ばれるもので、結核などの肉芽腫性疾患で見られる特徴です。この壊死物質は、細胞の構造が完全に失われた状態で、チーズ状の外観を示します。
- Caseous Necrosis(乾酪壊死):
- この壊死は肉眼的にチーズ状の外観をしているため、乾酪壊死と呼ばれます。特に結核などで頻繁に見られ、肉芽腫の中心部分に存在します。
細胞傷害、細胞死、適応
病理学の導入 (INTRODUCTION TO PATHOLOGY)
病理学の分野は、疾患の原因を理解し、疾患に関連する細胞、組織、臓器の変化を解明することに専念しています。これらの変化は患者に現れる症状や徴候の原因となります。病理学および医学を学ぶ中で学生が頻繁に目にする重要な用語が2つあります。
- 病因 (Etiology) とは、疾患の発症および進行の原因となる根本的な要因や修飾因子を指します。高血圧、糖尿病、癌など多くの一般的な疾患は、遺伝的な感受性とさまざまな環境要因の組み合わせで引き起こされることが明らかになっています。疾患の根底にある遺伝的および環境的要因の解明は、現代医学の主要なテーマです。
- 病態 (Pathogenesis) とは、疾患の発展および進行のメカニズムを指し、特定の疾患を特徴づける機能的および構造的異常を引き起こす細胞および分子の変化を説明します。したがって、病因 (etiology) は疾患が「なぜ」発生するのかを説明し、病態 (pathogenesis) は疾患が「どのように」進行するのかを説明します (図 2.1)。
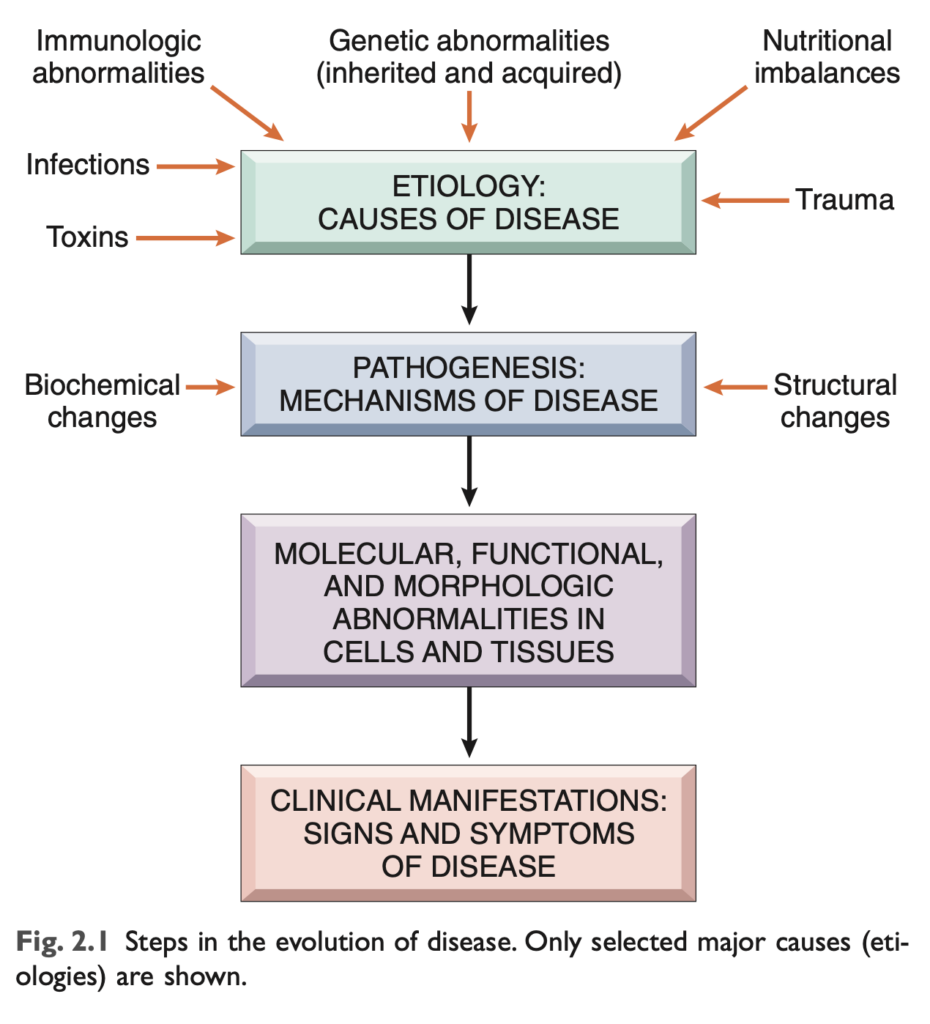
疾患の病因と病態を定義することは、疾患を理解するために不可欠であるだけでなく、合理的な治療法の開発や効果的な予防措置の基盤となります。このようにして、病理学は医学の実践のための科学的基盤を提供します。
臨床実践において診断を下し、治療を導くために、病理学者は細胞や組織の肉眼的または顕微鏡的な外観 (形態学、morphology) の変化、および体液 (血液や尿など) における生化学的変化を特定します。病理学者はまた、細胞、組織、臓器が損傷に反応して起こる生化学的、構造的、機能的な変化を定義するために、さまざまな形態学的、分子的、およびその他の技術を使用します。この章では、さまざまな内部 (遺伝的など) および外部 (環境的など) の異常やストレスによって引き起こされる細胞の異常について議論します。
ストレスと有害刺激に対する細胞の反応の概要 (OVERVIEW OF CELLULAR RESPONSES TO STRESS AND NOXIOUS STIMULI)
細胞は環境と積極的に相互作用し、変化する要求や外部からのストレスに適応するためにその構造と機能を常に調整しています。細胞内環境は通常厳密に調整されており、一定に保たれている状態は恒常性 (homeostasis) と呼ばれます。細胞が生理的ストレス (心臓における負荷の増加など) や有害な状態 (栄養不足など) に直面すると、適応して新たな安定状態に達し、存続と機能を維持します。しかし、適応能力が限界を超えるか、外部のストレスが本質的に有害または過剰である場合、細胞傷害が発生します (図 2.2)。一定の限界内では、損傷は可逆的であり、細胞は安定した基準状態に戻ります。しかし、ストレスが重度、持続的、または急速に発生すると、不可逆的な損傷と細胞死が引き起こされます。細胞死は、あらゆる組織や臓器における疾患の進展において最も重要なイベントの1つです。これには、虚血 (血流不足)、感染症、毒素、免疫反応など多様な原因が含まれます。細胞死はまた、胚発生、臓器の発達、組織の恒常性維持においても正常で不可欠なプロセスです。
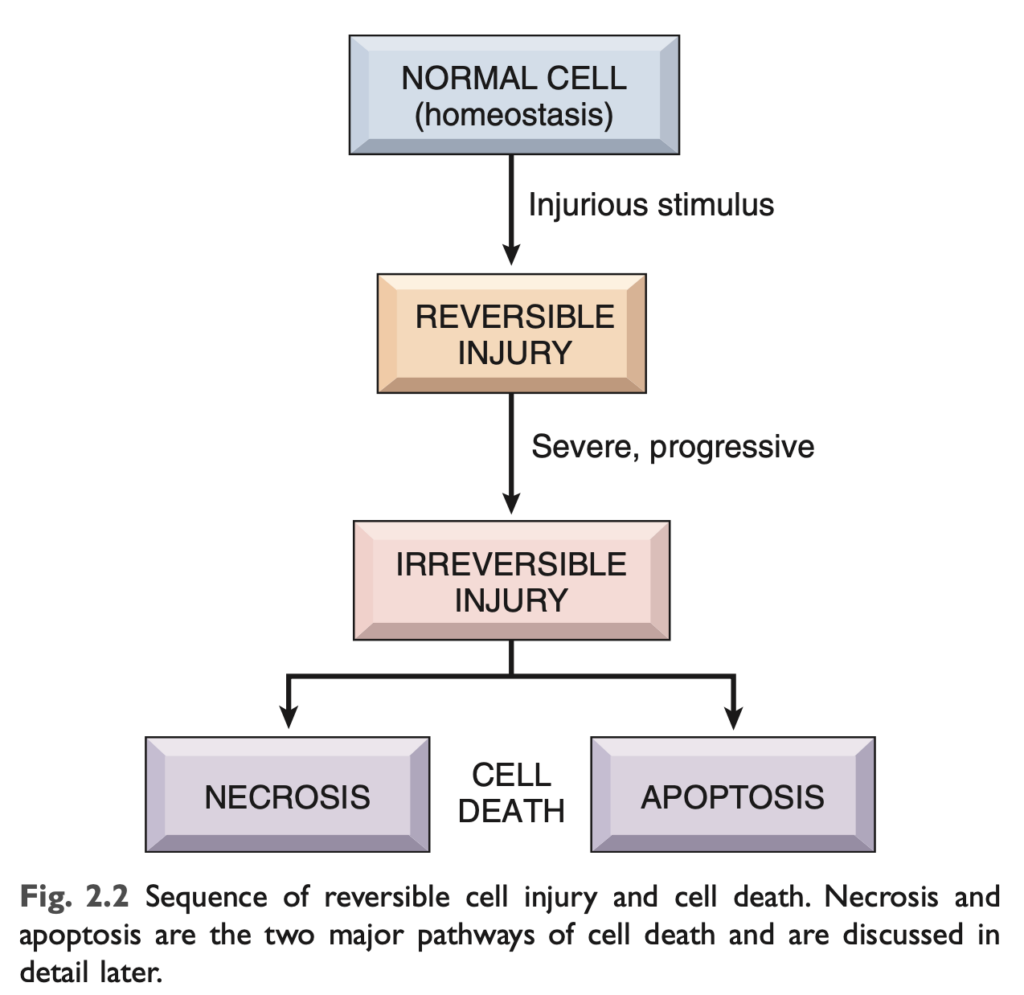
細胞への損傷はすべての疾患の基盤であるため、この章ではまず急性細胞傷害のさまざまな形態、可逆的損傷や細胞死の原因、メカニズム、および結果を考察します。次に、ストレスに対する細胞の適応を考え、最後に細胞や組織に影響を与えるその他の2つのプロセス、すなわち異常物質の沈着と細胞老化について議論します。
細胞傷害の原因 (CAUSES OF CELL INJURY)
細胞傷害の原因は、自動車事故による物理的外傷のような大規模なものから、特定の代謝疾患において機能しない酵素を引き起こす単一の遺伝子欠陥のようなものまで多岐にわたります。ほとんどの有害な刺激は、以下のカテゴリに分類されます。
- 低酸素症と虚血 (Hypoxia and ischemia)
低酸素症 (oxygen deficiency) は酸素不足を指し、虚血 (ischemia) は血液供給の減少を意味します。これらは細胞傷害の最も一般的な原因の1つです。両方とも組織に酸素を供給できなくなりますが、虚血はさらに必須栄養素の不足と有害代謝物の蓄積を引き起こします。低酸素症の最も一般的な原因は動脈閉塞に起因する虚血ですが、肺の疾患で血液の酸素化が不十分な場合や、貧血や一酸化炭素 (CO) 中毒によって血液の酸素運搬能力が低下することもあります。 - 毒素 (Toxins)
潜在的に有害な物質は、日常生活の中で環境中に存在しています。これには、大気汚染物質、殺虫剤、CO、アスベスト、タバコの煙、エタノール、薬物が含まれます。多くの薬物は、治療用の投与量であっても、感受性のある患者に細胞や組織の傷害を引き起こす可能性があり、過剰または不適切に使用された場合、さらに多くの人々に影響を与えることがあります (Chapter 7)。さらには、グルコース、塩分、水、酸素のような無害な物質であっても、有毒になることがあります。 - 感染性因子 (Infectious agents)
ウイルス、細菌、真菌、原虫などのあらゆるタイプの病原体が細胞を傷害します。これらの多様な因子による細胞傷害のメカニズムは、Chapter 9で議論されます。 - 免疫反応 (Immunologic reactions)
免疫系は病原体から体を防御しますが、免疫反応自体が細胞や組織を傷害することもあります。例として、自己の組織に対する自己免疫反応、環境物質に対するアレルギー反応、病原体に対する過剰または慢性的な免疫反応があります (Chapter 5)。これらすべての状況において、免疫反応は炎症反応を引き起こし、これが細胞や組織の損傷の原因となることが多いです。 - 遺伝的異常 (Genetic abnormalities)
遺伝的異常は、ダウン症候群に関連する先天性奇形のように顕著な病理学的変化を引き起こすこともあれば、鎌状赤血球貧血 (sickle cell anemia) におけるヘモグロビンの単一アミノ酸置換のように微細な変化を引き起こすこともあります (Chapter 7)。遺伝子の欠陥は、代謝異常 (inborn errors of metabolism) における酵素の欠如や、損傷したDNAまたは誤って折り畳まれたタンパク質の蓄積の結果として細胞傷害を引き起こします。これらが修復不能である場合、細胞死が引き起こされます。 - 栄養不均衡 (Nutritional imbalances)
貧困層におけるタンパク質-カロリー不足は依然として主要な細胞傷害の原因であり、ビタミン欠乏症も発展途上国のみならず、生活水準の高い先進国でも一般的です (Chapter 8)。逆に、過剰な食事摂取は肥満を引き起こし、2型糖尿病 (type 2 diabetes mellitus) や動脈硬化 (atherosclerosis) などの疾患の基盤となる重要な要因です。 - 物理的要因 (Physical agents)
外傷、極端な温度変化、放射線、電撃、急激な気圧変化などは、細胞に広範囲の影響を及ぼします (Chapter 8)。 - 老化 (Aging)
細胞老化 (cellular senescence) により、細胞がストレスに対応する能力が低下し、最終的には細胞および生体の死に至ります。細胞老化のメカニズムは、この章の最後に議論されています。
細胞傷害および細胞死の過程 (SEQUENCE OF EVENTS IN CELL INJURY AND CELL DEATH)
さまざまな有害な刺激は多様な生化学的メカニズムを通じて細胞を損傷しますが、ほとんどの細胞タイプにおいて、形態的および構造的な変化のステレオタイプ的な順序を引き起こします。
可逆的な細胞傷害 (Reversible Cell Injury)
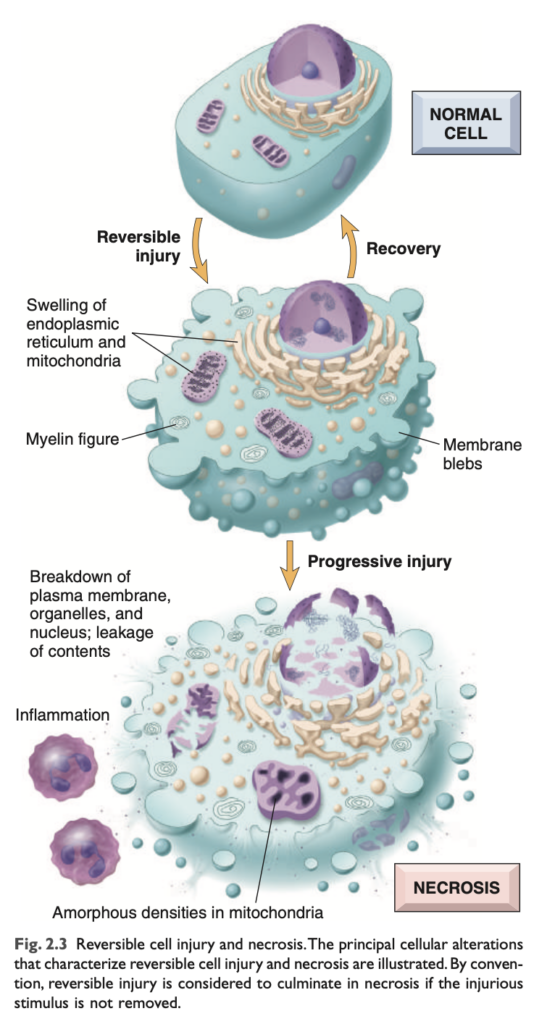
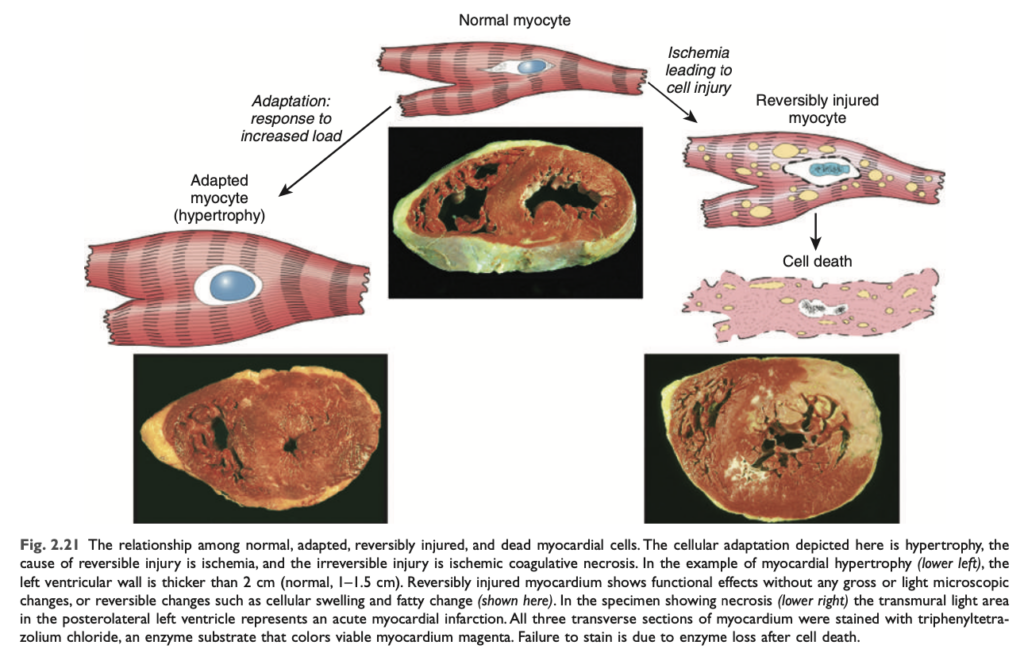
可逆的な傷害は、損傷を引き起こした刺激が除去された場合、損傷した細胞の機能および形態が正常に戻る段階です (図 2.3)。可逆的な損傷では、細胞や細胞内小器官が通常膨張します。これは、細胞膜のエネルギー依存性イオンポンプの機能不全により水分を取り込み、イオンおよび体液の恒常性を維持できなくなるためです。いくつかの形態の傷害では、損傷した細胞内に変性した小器官や脂質が蓄積することがあります。
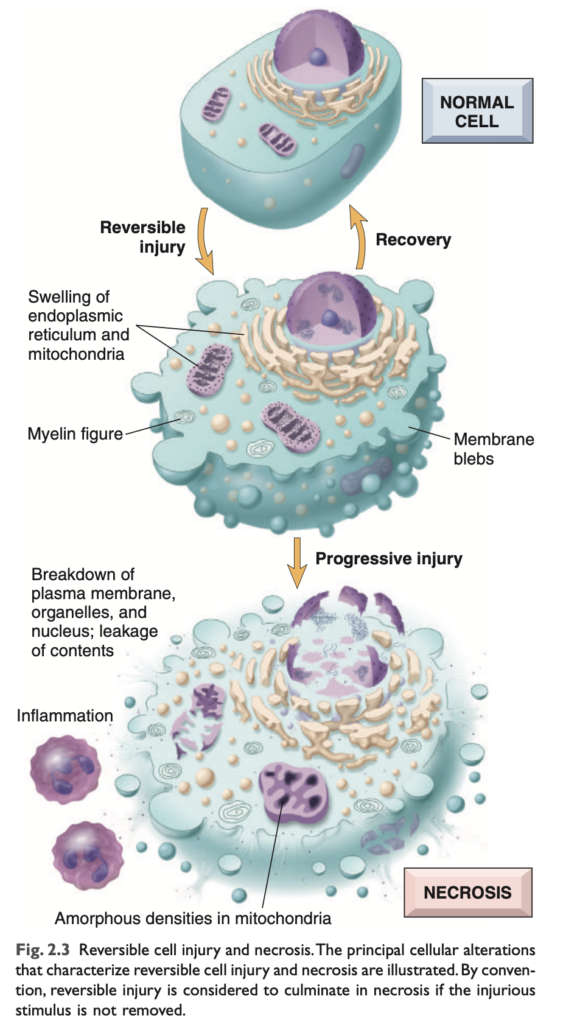
一部の状況では、有害な刺激により小器官 (特に小胞体) に特定の変化が誘発されることがあります。滑面小胞体はさまざまな化学物質の代謝に関与しており、これらの化学物質に曝露された細胞は適応応答として滑面小胞体の肥大 (hypertrophy) を示し、これが重要な機能的影響をもたらすことがあります。たとえば、多くの薬物 (バルビツレートなど) は、滑面小胞体にあるシトクロムP-450混合機能酸化酵素系 (cytochrome P-450 mixed-function oxidase system) によって肝臓で代謝されます。バルビツレートの長期使用は耐性状態を引き起こし、同じ効果を得るためにはより多くの薬物量が必要になります。この適応は、肝細胞の滑面小胞体の肥大およびP-450酵素活性の増加に起因します。P-450を介した化合物の修飾は、解毒につながることもあれば、危険な毒素に変換されることもあります。1つの例として、後述する四塩化炭素 (CCl4) が挙げられます。1つの薬物に適応した細胞は、同じ系によって処理される他の化合物の代謝能力も増加します。たとえば、フェノバルビタール (phenobarbital) を服用しているてんかん患者がアルコール摂取を増やした場合、滑面小胞体の肥大による応答として、抗てんかん薬の血中濃度が治療効果を下回るレベルにまで低下する可能性があります。
持続的または過度の有害な曝露が続くと、損傷した細胞は曖昧な「帰還不能点」を超えて細胞死に至ります。この移行点を定義することの臨床的重要性は明白です。細胞死を予測する生化学的および分子レベルの変化が特定できれば、可逆的な細胞傷害から不可逆的な細胞傷害への移行を防ぐための戦略を考案することが可能になるかもしれません。不可逆性を示す明確な形態学的または生化学的相関は存在しないものの、これには以下の3つの現象が関与しています。(1) ミトコンドリア機能 (酸化的リン酸化およびアデノシン三リン酸 [ATP] 生成) を元に戻せない、(2) 細胞膜および細胞内膜の構造および機能を失う、(3) DNA およびクロマチンの構造的完全性を失う。後述するように、リソソーム膜が損傷を受けると、損傷した細胞の酵素的な溶解が生じ、これが壊死 (necrosis) の最終段階です。
形態学的変化 (MORPHOLOGY)
可逆的細胞傷害の2つの主要な形態学的相関は、細胞の膨張 (cellular swelling) と脂肪変性 (fatty change) です。
- 細胞の膨張 (Cellular swelling): Fig. 2.4
細胞膜の透過性が増加する細胞傷害において一般的に見られます。光学顕微鏡では見分けが難しいことがありますが、臓器全体のレベルでは明らかになることが多いです。臓器内の多くの細胞が影響を受けると、毛細血管の圧迫によって蒼白 (pallor) が生じ、膨張 (turgor) が増加し、臓器の重量が増加します。顕微鏡的には、細胞質内に小さく透明な空胞 (vacuoles) が見られることがあり、これは拡張して切り離された小胞体 (endoplasmic reticulum, ER) の一部を表しています。この非致死的な損傷パターンは、水腫変性 (hydropic change) または空胞変性 (vacuolar degeneration) とも呼ばれます。 - 脂肪変性 (Fatty change):
細胞質内にトリグリセリド (triglyceride) を含む脂質の空胞が現れることによって示されます。これは主に肝臓 (liver) のような脂質代謝に関与する臓器で見られ、Chapter 16で詳しく説明されます。
損傷した細胞の細胞質 (cytoplasm) は、さらに赤みを帯び (エオジン好性, eosinophilic)、壊死へ進行するとこの変化がさらに顕著になります。他の細胞内の変化としては、(1) 細胞膜の変形 (plasma membrane alterations) には、膨隆 (blebbing)、鈍化 (blunting)、微絨毛の変形 (distortion of microvilli)、細胞間結合の緩み (loosening of intercellular attachments) があります。(2) ミトコンドリアの変化には、膨張 (swelling) およびリン脂質に富む無定形密度 (phospholipid-rich amorphous densities) の出現があります。(3) 小胞体の拡張およびリボソームの脱離、ポリソームの解離 (dissociation of polysomes)。(4) 核の変化には、クロマチンの凝集 (clumping of chromatin) があります。細胞質内には、損傷した細胞膜から派生した**「ミエリンフィギュア (myelin figures)**」と呼ばれる、ミエリン鞘に似たリン脂質の集合体が含まれることもあります。
細胞死 (Cell Death)
細胞が損傷を受けると、損傷の性質や重症度に応じてさまざまなメカニズムによって死に至ります。
- 重篤な障害 (Severe disturbances)、たとえば酸素や栄養供給の喪失や毒素の作用などは、急速かつ制御不能な「事故的細胞死 (accidental cell death)」という死の形態を引き起こします。事故的細胞死の形態学的表現は壊死 (necrosis) です (ギリシャ語で「死」を意味する necros) (表2.1)。壊死は、虚血、毒素への曝露、さまざまな感染症、外傷など一般的に見られる傷害において、主要な細胞死の経路です。壊死は伝統的に、修復不能な重度の損傷の最終的な結果と見なされ、特定のシグナルや生化学的メカニズムによって制御されるものではないと考えられています。言い換えれば、壊死は事故的に発生し、損傷が修復不可能なほど重度であるため、細胞の多くの構成要素が単純に機能しなくなったり、崩壊したりするからです。
- 一方で、損傷がそれほど深刻でない場合、あるいは正常なプロセス中に細胞が除去される必要がある場合、細胞は一連の分子経路を活性化させ、死に至ります。このような細胞死は治療薬や遺伝子変異によって操作可能であるため、「制御された細胞死 (regulated cell death)」と呼ばれます。ほとんどの制御された細胞死の形態学的外観はアポトーシス (apoptosis) です (表2.1参照)。場合によっては、制御された細胞死は壊死とアポトーシスの両方の特徴を示し、「ネクロプトーシス (necroptosis)」と呼ばれます。以前は認識されていなかったこれらの細胞死の形態が、特定の遺伝子やシグナル伝達経路によって制御されていることが判明したことで、細胞死が制御可能なプロセスであることが示されました。制御された細胞死という概念は、病的状態における細胞の喪失を防ぐために特定の分子経路を治療的に標的にできる可能性をも示唆しています。アポトーシスは、さまざまな内在的異常を持つ細胞を排除し、細胞の断片が炎症反応を引き起こすことなく除去されるプロセスです。この「クリーンな」形態の細胞自殺は、細胞のDNAやタンパク質が修復不可能なほど損傷を受けた場合や、必要な生存シグナルを奪われた場合に病的状況で発生します。しかし、壊死とは異なり、アポトーシスは必ずしも病的プロセスを示すものではなく、正常な発生過程中に不要な細胞を除去し、一定の細胞数を維持する役割を果たします。これらのタイプの生理的な細胞死は「プログラムされた細胞死 (programmed cell death)」とも呼ばれます。
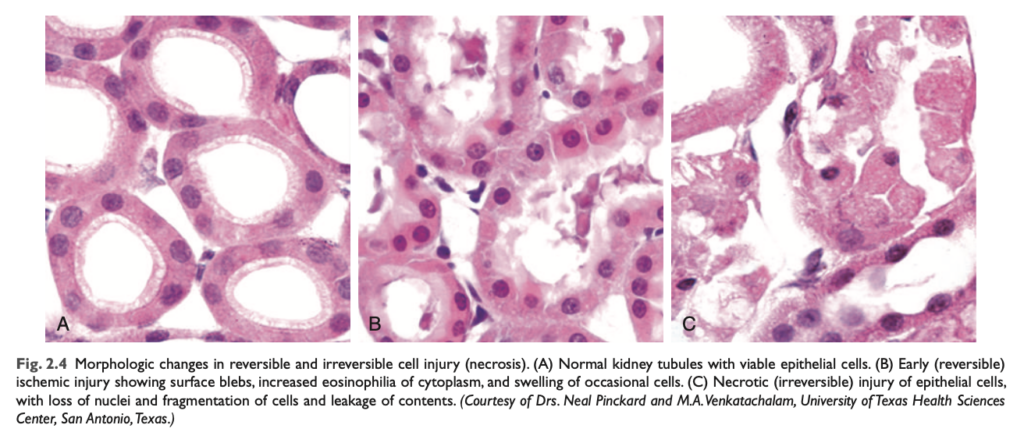
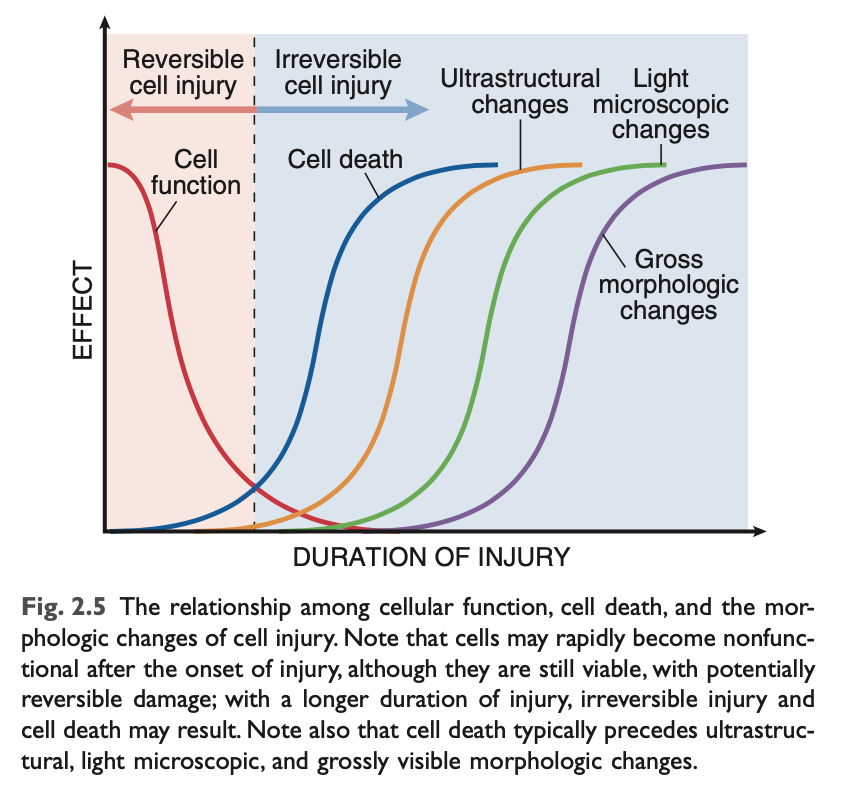
細胞の機能は、細胞が死ぬ前に失われることが多く、細胞傷害や死の形態学的変化は機能や生存能力の喪失からかなり遅れて現れます (図2.5)。たとえば、心筋細胞は虚血後1〜2分で収縮しなくなりますが、虚血が20〜30分経過しないと死に至りません。虚血性筋細胞の死を示す形態学的特徴は、細胞死後2〜3時間以内に電子顕微鏡で確認できますが、光学顕微鏡では6〜12時間後になって初めて明らかになります。
壊死 (Necrosis)
壊死は、細胞膜が崩壊し、細胞内酵素が漏れ出して最終的に細胞を消化する細胞死の一形態です (図2.3)。壊死は、死細胞から放出された物質によって誘発される局所的な宿主反応である炎症を引き起こし、この炎症は破片を除去し、修復プロセスを開始するために役立ちます (Chapter 3)。細胞を消化するための酵素は、死にかけている細胞自身や、炎症反応の一環として動員された白血球から由来することがあります。壊死は、可逆的な細胞傷害が修復できなくなった結果として生じることが多いです。
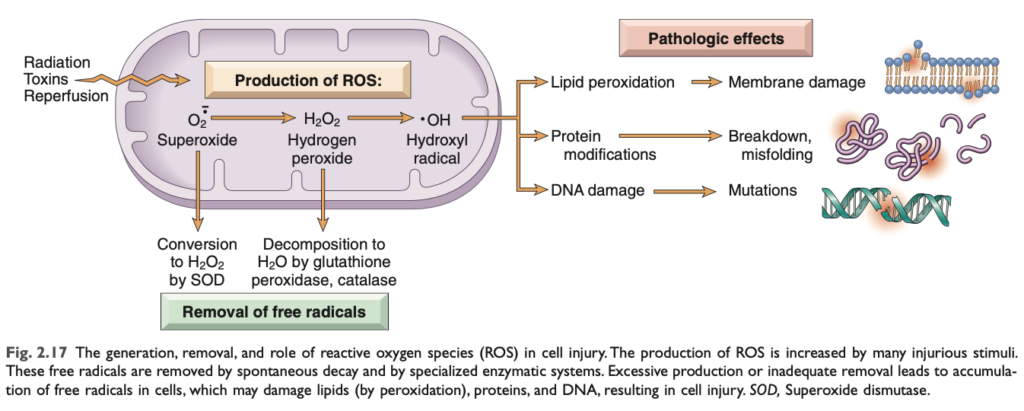
壊死の生化学的メカニズムは、さまざまな有害刺激によって異なります。これらのメカニズムには、酸素供給の減少やミトコンドリアの損傷によるATP生成の失敗、細胞膜やリソソーム膜を含む細胞膜の損傷 (細胞内容物や酵素の漏出を引き起こす)、および活性酸素種 (ROS) による細胞内脂質、タンパク質、核酸の不可逆的な損傷などが含まれます。これらの生化学的メカニズムについては、後の章で個別に議論します。
形態学的変化 (MORPHOLOGY)
壊死は、損傷した細胞の細胞質 (cytoplasm) と核 (nuclei) の変化によって特徴づけられます (図2.3および2.4C)。
- 細胞質の変化 (Cytoplasmic changes):
壊死細胞はエオジン好性 (eosinophilia) が増加し、これはエオジン (ヘマトキシリンおよびエオジン染色 [H&E] の「E」) によって赤く染色されることに起因します。これは部分的には変性した細胞質タンパク質へのエオジンの結合が増加するためであり、部分的には細胞質内の塩基好性 (basophilic) RNA が失われるためです (塩基好性は青色染料であるヘマトキシリン [H&E の「H」] への結合によるものです)。生細胞と比較して、壊死細胞はガラスのような均質な外観を持つことがあり、これは軽く染色されるグリコーゲン粒子の喪失によるものです。壊死細胞では、可逆的損傷を受けた細胞よりもミエリンフィギュア (myelin figures) が目立ちます。酵素が細胞質小器官を消化すると、細胞質は空胞化し、「虫食い状」に見えることがあります。電子顕微鏡では、壊死細胞は細胞膜や小器官膜の不連続性、大きな無定形のミトコンドリア内密度の出現を伴うミトコンドリアの著しい膨張、リソソームの破壊、および細胞質内のミエリンフィギュアによって特徴づけられます。 - 核の変化 (Nuclear changes):
核の変化は、DNAとクロマチンの分解によって引き起こされる3つのパターンのいずれかを示します。核縮小 (pyknosis) は核の縮小および塩基好性の増加を特徴とし、DNAが暗く縮んだ塊に凝縮されます。ピクノティックな核は、断片化 (karyorrhexis) を起こすことがあります。最終的に核は融解 (karyolysis) を起こし、DNAのデオキシリボヌクレアーゼ (DNase) 活性による消化によって塩基好性が消失します。1〜2日後には、死細胞の核は完全に消失することがあります。 - 壊死細胞の運命 (Fates of necrotic cells):
壊死細胞はしばらく存在するか、酵素によって消化されて消失します。死細胞は、他の細胞によって貪食されるか、さらに脂肪酸に分解されるミエリンフィギュアによって置き換えられることがあります。これらの脂肪酸はカルシウム塩と結合し、死細胞が最終的に石灰化することがあります。
壊死のほとんどのタイプは、明確な肉眼的外観を持っています。ただし、フィブリノイド壊死 (fibrinoid necrosis) は組織学的検査によってのみ検出されます。
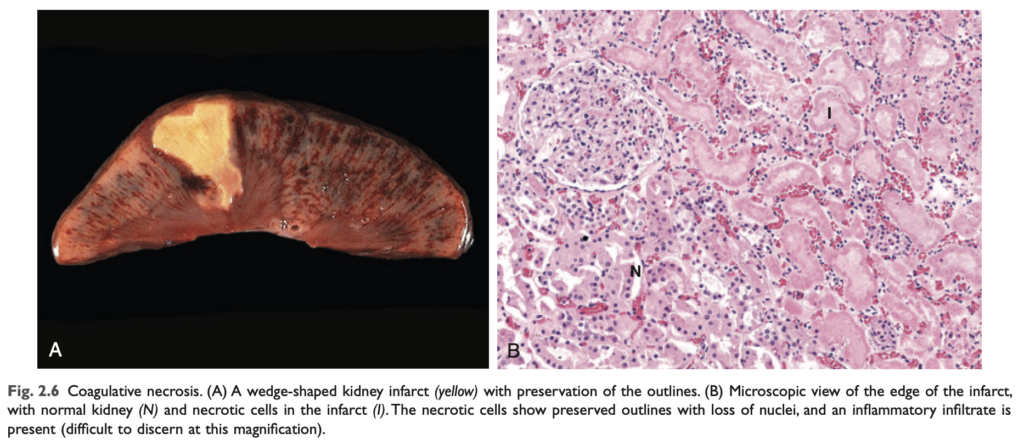
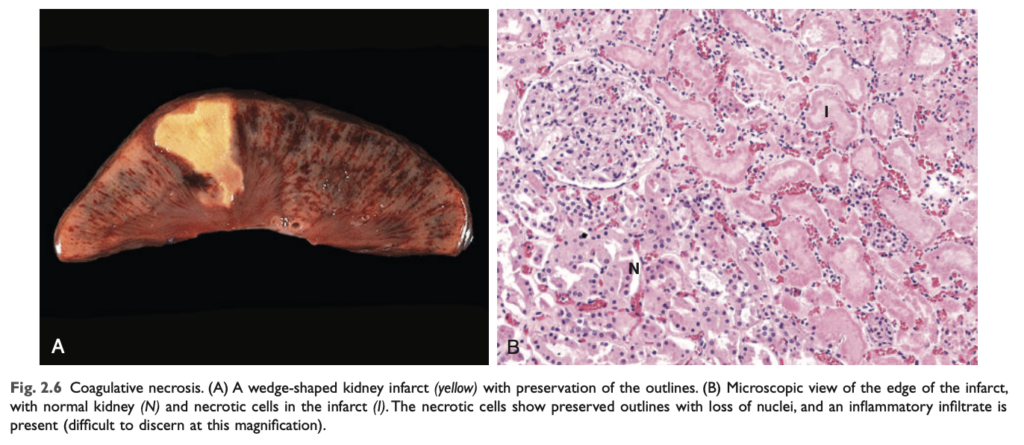
- 凝固壊死 (Coagulative necrosis):
凝固壊死では、細胞が死んだ後も数日間、基礎的な組織構造が保存されます (図2.6)。影響を受けた組織は硬い質感を帯びます。損傷により構造タンパク質だけでなく酵素も変性し、死細胞のプロテオリシス (タンパク質分解) が抑制されるため、エオジン好性で核を持たない細胞が数日から数週間にわたって残存します。壊死の部位には白血球が集まり、死細胞は白血球のリソソーム酵素の作用によって最終的に消化されます。その後、細胞破片は主に浸潤する好中球やマクロファージによって貪食され、除去されます。凝固壊死は、脳を除くすべての固体臓器において虚血によって引き起こされる梗塞 (infarcts) に特徴的です。
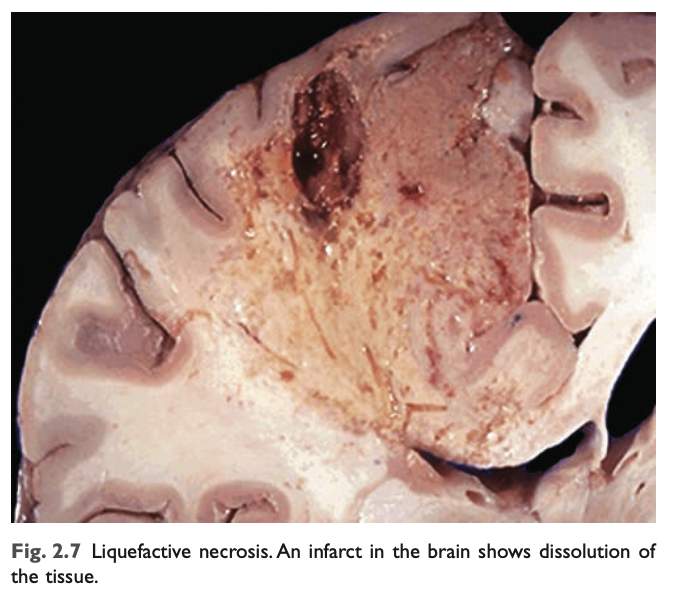
- 液化壊死 (Liquefactive necrosis):
液化壊死は、局所的な細菌感染症や、まれに真菌感染症において見られます。これらの微生物は炎症細胞の急速な蓄積を刺激し、白血球の酵素が組織を消化 (「液化」) します。特定の理由は不明ですが、中枢神経系の細胞が低酸素症で死ぬと、液化壊死がしばしば引き起こされます (図2.7)。いずれにせよ、死細胞は完全に消化され、組織は粘性の液体に変わり、最終的には貪食細胞によって除去されます。急性炎症 (たとえば細菌感染症) によってこのプロセスが開始された場合、生成される物質はしばしば黄色いクリーム状になり、膿 (pus) と呼ばれます (Chapter 3)。
- 壊疽性壊死 (Gangrenous necrosis):
壊疽性壊死は明確な細胞死のパターンではありませんが、この用語は臨床実践でよく使われます。通常、血液供給を失い、複数の組織層に凝固壊死が生じた四肢 (一般的には下肢) の状態を指します。細菌感染が加わると、細菌や白血球による組織破壊のため、形態学的外観は液化壊死に変わります (「湿性壊疽 (wet gangrene)」)。 - 乾酪壊死 (Caseous necrosis):
乾酪壊死は結核感染巣で最も頻繁に見られます。乾酪とは「チーズのような」という意味で、壊死部位が黄色白色で脆い外観をしていることを指します (図2.8)。顕微鏡的には、壊死巣は断片化または溶解した細胞の集まりとして現れ、H&E染色組織切片ではピンク色の顆粒状の無定形物質として見られます。凝固壊死とは異なり、組織構造は完全に破壊され、細胞の輪郭は識別できません。乾酪壊死は、マクロファージや他の炎症細胞の集まりに囲まれており、この外観は肉芽腫性炎症病変 (granuloma) と呼ばれます (Chapter 3)。

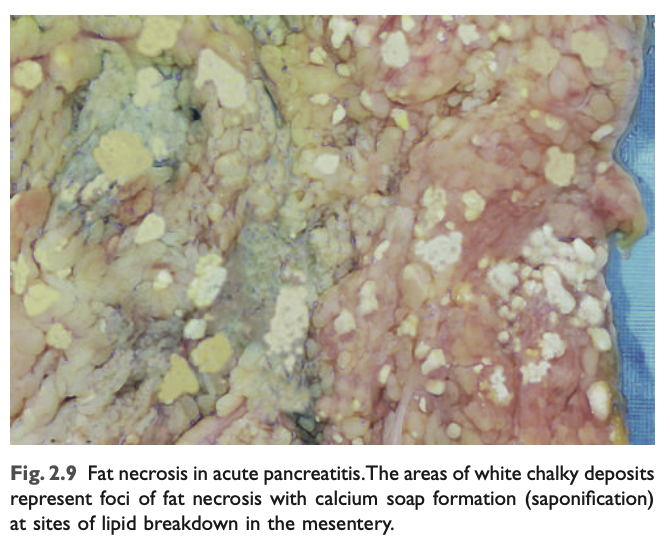
- 脂肪壊死 (Fat necrosis):
脂肪壊死は、膵臓や腹膜腔の脂肪組織が破壊される局所的な領域を指し、急性膵炎 (acute pancreatitis) で見られます (Chapter 17)。この疾患では、膵酵素がアシナ細胞や管から漏れ出し、腹膜内の脂肪細胞の膜を液化させ、リパーゼが脂肪細胞内のトリグリセリドエステルを分解します。遊離された脂肪酸はカルシウムと結合し、外科医や病理学者が病変を識別できるような白くチョーク状の領域 (脂肪鹸化, fat saponification) を形成します (図2.9)。組織学的には、壊死部位には壊死脂肪細胞の影があり、カルシウム沈着や炎症反応に囲まれています。
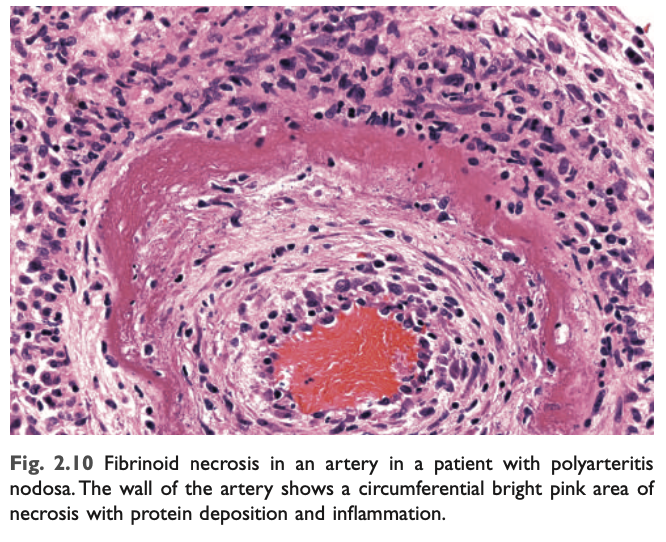
- フィブリノイド壊死 (Fibrinoid necrosis):
フィブリノイド壊死は特別な形態の壊死であり、通常は抗原と抗体の複合体が血管壁に沈着する免疫反応で見られますが、重度の高血圧でも発生することがあります。損傷を受けた血管壁に沈着した免疫複合体と漏出した血漿タンパク質は、H&E染色標本で鮮やかなピンク色の無定形物質として見られ、病理学者はこれをフィブリノイド (フィブリン様) と呼びます (図2.10)。ポリアルテリティス・ノドーサ (polyarteritis nodosa) のような免疫介在疾患においてこのタイプの壊死が見られます (Chapter 5)。
細胞膜の損傷によるタンパク質の漏出 (Leakage of intracellular proteins)
損傷した細胞膜を通して細胞内タンパク質が漏れ出し、最終的には血液循環に入ることで、血液や血清サンプルを使用して組織特異的な壊死を検出する手段が提供されます。たとえば、心筋にはクレアチンキナーゼ (creatine kinase) や収縮性タンパク質のトロポニン (troponin) の特有のアイソフォームが含まれており、一方で、肝胆管上皮細胞にはアルカリホスファターゼ (alkaline phosphatase)、肝細胞にはトランスアミナーゼ (transaminases) が含まれています。これらの組織に不可逆的な損傷や細胞死が生じると、これらのタンパク質の血清レベルが上昇し、臨床的に組織損傷の指標として有用です。
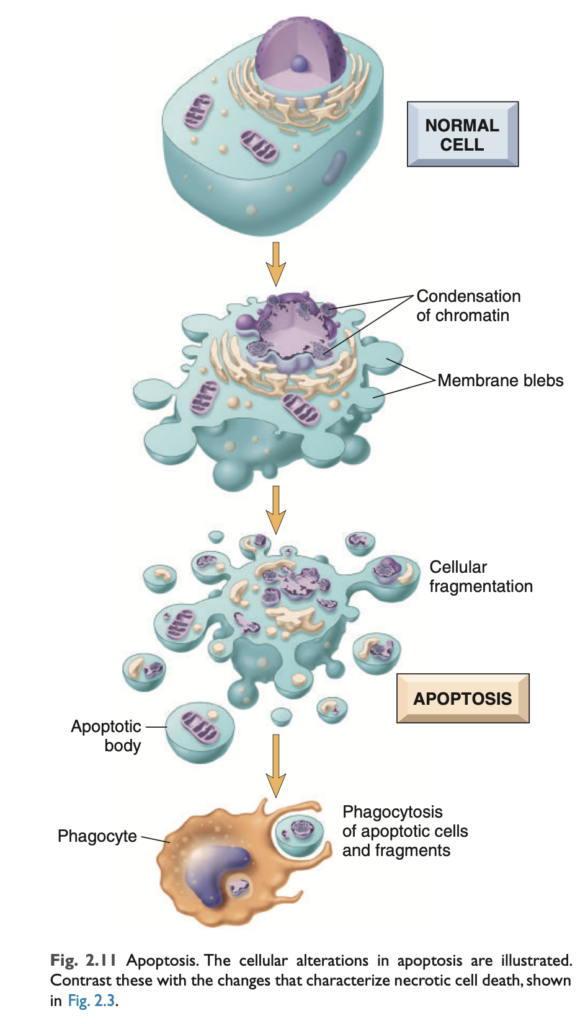
アポトーシス (Apoptosis)
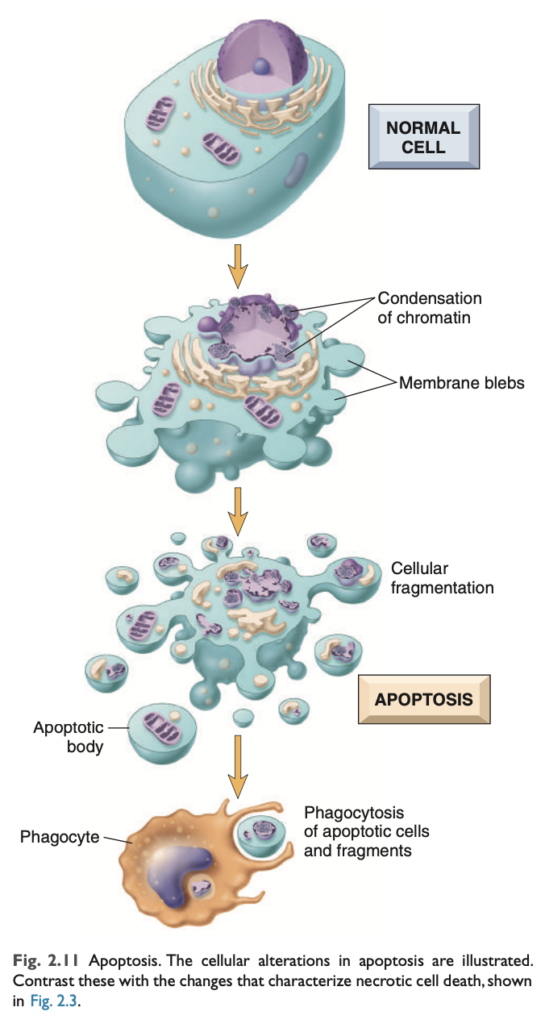
アポトーシスは、細胞が自身の核DNA (nuclear DNA) および核・細胞質タンパク質 (nuclear and cytoplasmic proteins) を分解する酵素を活性化する細胞死の経路です (図2.11)。アポトーシス細胞の断片が外れることで、その名前の由来である「脱落 (falling off)」の外観が現れます。アポトーシス細胞の細胞膜 (plasma membrane) は保持されますが、その膜は変化し、断片 (アポトーシス小体, apoptotic bodies) は非常に「食べやすく」なり、速やかに食細胞 (phagocytes) によって処理されます。死んだ細胞およびその断片は、細胞内容物の漏出がほとんどなく除去されるため、アポトーシスによる細胞死は炎症反応を引き起こしません。このため、アポトーシスは壊死とは多くの点で異なります (表2.1)。
アポトーシスの原因 (Causes of Apoptosis)
アポトーシスは多くの正常な状況で発生し、有害な細胞や役割を終えた細胞を排除する役割を果たします (表2.2)。また、特に細胞のDNA やタンパク質が損傷している場合、病的なイベントとしても発生し、修復不能な細胞が除去されます。
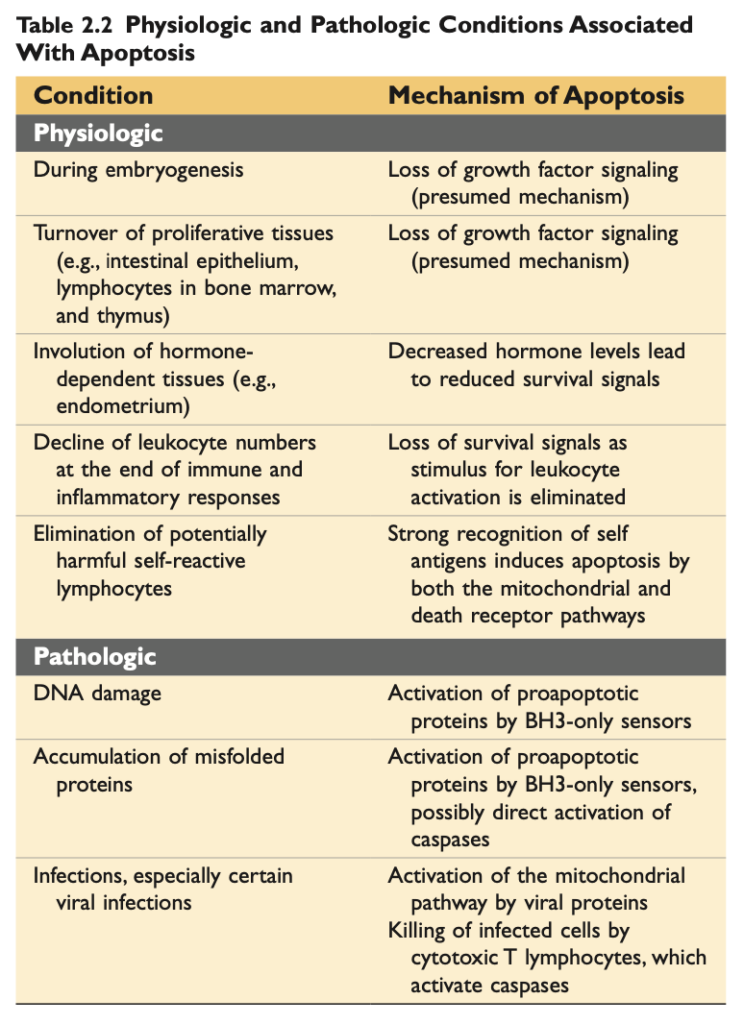
- 生理的アポトーシス (Physiologic apoptosis): 生物の正常な発生過程では、いくつかの細胞が死に、新しい細胞に置き換えられます。成熟した生物では、増殖性の高い組織 やホルモン反応性組織が増殖と細胞損失のサイクルを経験します。このような場合、細胞死は常にアポトーシスによって行われ、不要な細胞が炎症を引き起こすことなく除去されます。免疫系では、アポトーシスは免疫反応の終結後に残った余分な白血球 (leukocytes) や、自己抗原を認識し、自己免疫疾患を引き起こす可能性のあるリンパ球 (lymphocytes) を排除します。
アポトーシスのメカニズム (Mechanisms of Apoptosis)
アポトーシスは、細胞死と生存のシグナルのバランスを制御し、最終的にはカスパーゼ (caspases) と呼ばれる酵素の活性化を制御する生化学的経路によって規制されています。カスパーゼは、システインプロテアーゼ (cysteine proteases) であり、アスパラギン酸残基 (aspartic acid residues) の後にタンパク質を切断します。2つの異なる経路がカスパーゼの活性化に収束します。それは、ミトコンドリア経路 (mitochondrial pathway) とデスレセプター経路 (death receptor pathway) です (図2.12)。これらの経路は交差することもありますが、通常は異なる条件下で誘導され、異なる分子を含み、生理学的および病理学的役割が異なります。アポトーシス細胞死の最終的な結果は、食細胞によるアポトーシス小体の除去です。
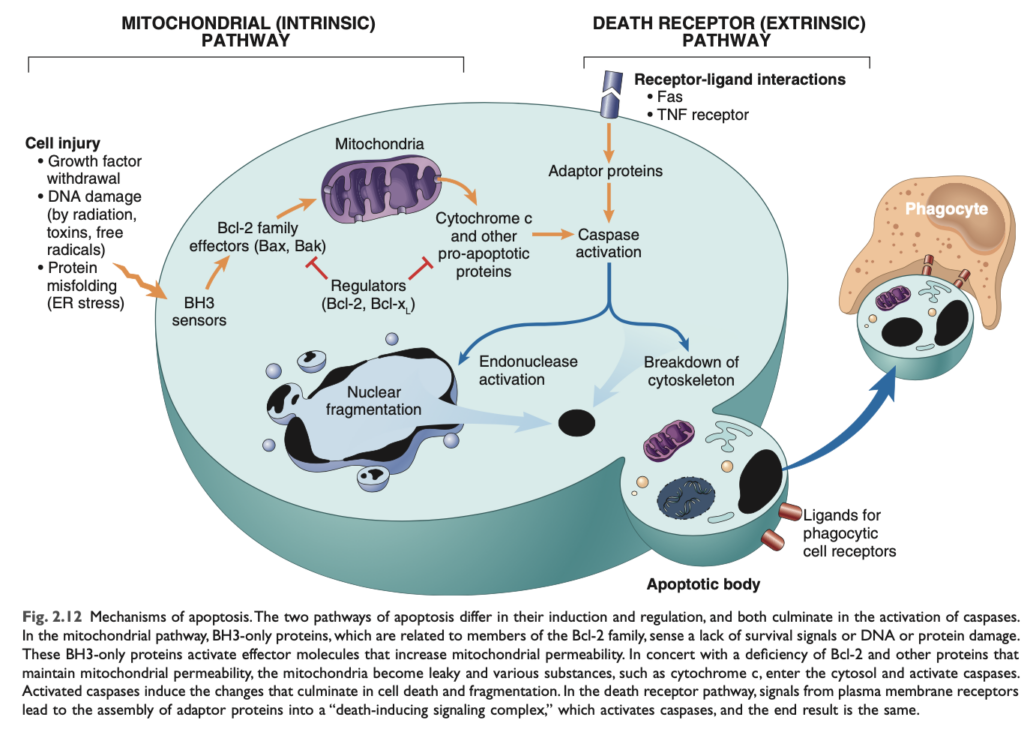
- ミトコンドリア経路 (Mitochondrial pathway):
ミトコンドリア経路は、ほとんどの生理的および病理学的状況でアポトーシスを引き起こすと考えられています。ミトコンドリアにはシトクロムC (cytochrome c) を含む、アポトーシスを誘導する能力を持ついくつかのタンパク質が含まれています。ミトコンドリア膜が透過性になると、シトクロムCが細胞質に漏れ出し、カスパーゼの活性化とアポトーシス死を引き起こします。Bcl-2 を代表とする20種類以上のタンパク質のファミリーがミトコンドリアの透過性を制御します。健康な細胞では、Bcl-2および関連タンパク質Bcl-xLが、成長因子や他の刺激に応答して産生され、ミトコンドリア膜の完全性を維持しますが、これは主にBaxおよびBakという2つのプロアポトーシスタンパク質 (proapoptotic proteins) を抑制することによって行われます。細胞が成長因子や生存シグナルを奪われたり、DNAを損傷させる物質にさらされたり、誤ったタンパク質が許容できない量蓄積すると、一部のBH3タンパク質 (BH3 proteins) が活性化されます。これらのセンサーは生命維持バランスをプロアポトーシスタンパク質であるBakおよびBaxの側に傾けます。その結果、BakおよびBaxが二量体化し、ミトコンドリア膜に挿入され、シトクロムCや他のミトコンドリアタンパク質が細胞質に流出するチャネルを形成します。シトクロムCが細胞質に入ると、それは特定の補因子と共にカスパーゼ-9 (caspase-9) を活性化します。最終的な結果はカスパーゼカスケードの活性化であり、最終的には核の断片化 (nuclear fragmentation) とアポトーシス小体の形成につながります。 - デスレセプター経路 (Death receptor pathway):
多くの細胞はデスレセプター (death receptors) と呼ばれる表面分子を発現しており、これがアポトーシスを引き起こします。これらのほとんどは、腫瘍壊死因子 (TNF) 受容体ファミリー (TNF receptor family) のメンバーであり、その細胞質領域には「デスドメイン (death domain)」と呼ばれる保存された領域が含まれています。このデスドメインは細胞死に関与する他のタンパク質との相互作用を媒介します。代表的なデスレセプターはTNF受容体1型 (type I TNF receptor) およびFas (CD95) です。Fasリガンド (FasL) は、主に活性化されたTリンパ球に発現する膜タンパク質です。これらのT細胞がFasを発現する標的を認識すると、Fas分子はFasLによって交差連結され、デスドメインを介してアダプタタンパク質と結合します。これによりカスパーゼ-8 (caspase-8) が動員されて活性化され、さらに下流のカスパーゼを活性化します。デスレセプター経路は、自己反応性リンパ球 (self-reactive lymphocytes) の排除や、一部の細胞傷害性Tリンパ球 (CTLs) による標的細胞の殺害に関与しています。
どちらの経路でも、カスパーゼ-9またはカスパーゼ-8が活性化された後、それは他のカスパーゼを切断して活性化し、最終的には細胞のタンパク質や核を分解する酵素を活性化します。最終的な結果は、アポトーシスの特徴的な細胞断片化です。
アポトーシス細胞の除去 (Clearance of apoptotic cells)
アポトーシス細胞およびその断片は、いくつかの「食べてください (eat-me)」シグナルを発して食細胞を引き寄せます。たとえば、正常な細胞ではホスファチジルセリン (phosphatidylserine) が細胞膜の内側のリーフレットに存在しますが、アポトーシス細胞ではこのリン脂質 (phospholipid) が外側のリーフレットに「反転 (flips)」し、これが組織マクロファージ (tissue macrophages) に認識され、アポトーシス細胞の貪食につながります。アポトーシスで死にかけている細胞はまた、食細胞を呼び寄せる可溶性因子も分泌します。細胞膜の変化や分泌されるタンパク質は、細胞が膜損傷を受けて内容物を放出する前に、死細胞を迅速に除去するための役割を果たします (これは炎症を引き起こす可能性があります)。食細胞によるアポトーシス細胞の貪食は非常に効率的であり、死細胞は痕跡を残すことなく消失し、炎症はほとんど見られません。
形態学的変化 (MORPHOLOGY)
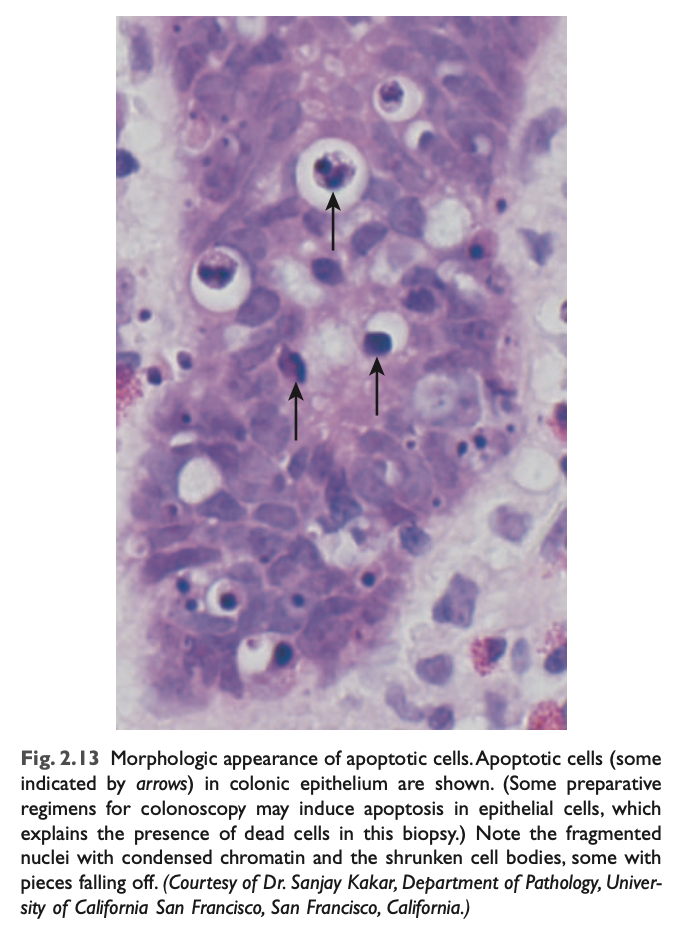
H&E染色された組織切片では、アポトーシス細胞の核はクロマチンの凝縮と集積のさまざまな段階を示し、最終的には核断片化 (karyorrhexis) に至ります (図2.13)。分子レベルでは、これはヌクレオソームサイズの断片 (nucleosome-sized fragments) へのDNAの断片化として反映されます。細胞は急速に縮小し、細胞質の芽を形成し、細胞質および小器官の膜結合片で構成されたアポトーシス小体に断片化されます (図2.11)。これらの断片は速やかに排出され、炎症反応を引き起こすことなく食細胞によって貪食されるため、たとえ大量のアポトーシスが発生しても、組織学的にはほとんど検出できない場合があります。
その他の細胞死の経路 (Other Pathways of Cell Death)
壊死とアポトーシスに加えて、異なる特徴を持つ2つの細胞死パターンが報告されています。これらの経路の疾患における重要性はまだ確立されていませんが、現在の研究の対象であり、基本的な概念を理解しておくことが有益です。
- ネクロプトーシス (Necroptosis):
この細胞死は、TNF受容体 (TNF receptors) や他の不明確なトリガーの関与によって開始されます。アポトーシスの外因性経路もTNF受容体に下流で連動しますが、ネクロプトーシスではレセプター相互作用タンパク質キナーゼ (RIP kinases) と呼ばれるキナーゼが活性化され、壊死に類似した一連の細胞崩壊イベントが引き起こされます。ネクロプトーシスという名前は、壊死とアポトーシスの両方の特徴を含むことを意味しています。一部の感染症はこの経路を介して細胞を殺すと考えられており、虚血性傷害やその他の病的状況、特にTNF を産生する炎症反応に関連する病態で役割を果たすと仮定されています。しかし、いつ、なぜこれが発生するのか、人間の疾患におけるその重要性は十分に理解されていません。 - パイロプトーシス (Pyroptosis):
この細胞死は、インフラマソーム (inflammasome) と呼ばれる細胞質の危険感知タンパク質複合体の活性化に関連しています (Chapter 5)。インフラマソームの活性化の最終結果はカスパーゼ (caspases) の活性化であり、これにより炎症を引き起こすサイトカイン (cytokines) の産生が誘導されます。炎症はしばしば発熱として現れ、他のカスパーゼがアポトーシスを引き起こします。このため、アポトーシスと炎症が共存します。パイロプトーシスという名前は、アポトーシスと発熱 (ギリシャ語のpyro = 火) の関連に由来しています。この経路は、一部の感染性微生物が感染細胞の死を引き起こすメカニズムの1つと考えられていますが、他の病的状況における役割は不明です。
オートファジー (Autophagy)
オートファジー(「自己食作用」)とは、細胞が自身の構成要素をリソソームによって消化するプロセスを指します。これは栄養不足時に細胞が生存するためのメカニズムであり、飢えた細胞が自身の内容物を食べ、それを再利用して栄養素やエネルギーを供給します。このプロセスでは、細胞内小器官や細胞質の一部がまず小胞体由来のオートファジー空胞 (autophagic vacuole) に隔離され、その形成は栄養不足を感知する細胞質タンパク質によって開始されます (図2.14)。その後、空胞はリソソームと融合してオートファゴリソソーム (autophagolysosome) を形成し、そこでリソソーム酵素が細胞成分を消化します。特定の状況下では、オートファジーは組織の萎縮と関連しており、困難な時期に細胞が生存するための適応を表している可能性があります。しかし、飢えた細胞が内容物を食べるだけでは対処できなくなると、最終的にアポトーシスによる細胞死に至ることがあります。オートファジーは虚血性損傷や一部の筋疾患で広く見られます。オートファジーに関与する遺伝子の多型は、炎症性腸疾患と関連していますが、オートファジーと腸の炎症との間の機構的な関連は不明です。がんにおけるオートファジーの役割についてはChapter 6で議論されます。このように、かつてあまり注目されなかった細胞内の生存経路が、人間の病気において広範な役割を果たす可能性があることが示唆されています。

要約 (SUMMARY)
細胞傷害および細胞死のパターン (Patterns of Cell Injury and Cell Death)
- 可逆的な細胞傷害 (Reversible cell injury): 細胞の膨張、脂肪変性、細胞膜の膨隆および微絨毛の喪失、ミトコンドリアの膨張、小胞体 (ER) の拡張、エオジン好性 (eosinophilia) の増加 (細胞質RNAの減少による)
- 壊死 (Necrosis): 細胞質エオジン好性の増加、核の縮小、断片化および溶解、細胞膜および小器官膜の崩壊、ミエリンフィギュアの豊富な存在、細胞内容物の漏出と酵素的消化を伴う事故的細胞死
- 壊死の形態学的タイプ (Morphologic types of tissue necrosis): さまざまな状況下で、組織の壊死は以下の特定のパターンを示すことがある: 凝固壊死 (coagulative)、液化壊死 (liquefactive)、壊疽壊死 (gangrenous)、乾酪壊死 (caseous)、脂肪壊死 (fat)、およびフィブリノイド壊死 (fibrinoid)
- アポトーシス (Apoptosis): 不要で修復不能な細胞をホストの反応を最小限に抑えて除去するための制御された細胞死のメカニズム。これはカスパーゼ (caspases) によって開始され、タンパク質やDNAの酵素的分解を伴い、死細胞は食細胞によって迅速に認識され、除去される。
- アポトーシスは2つの主要な経路によって開始される:
- ミトコンドリア経路 (Mitochondrial pathway): 生存シグナルの喪失、DNA損傷、および誤ったタンパク質の蓄積 (小胞体ストレス) によって引き起こされる。これはミトコンドリア膜から細胞質へのプロアポトーシスタンパク質 (proapoptotic proteins) の漏出に関連し、カスパーゼ活性化を引き起こす。これには、生存シグナル (成長因子など) によって誘導されるBclファミリー (Bcl family) の抗アポトーシスタンパク質が関与している。
- デスレセプター経路 (Death receptor pathway): CTLs による自己反応性リンパ球の除去や損傷に関与しており、隣接する細胞上のリガンド (ligands) によるTNF受容体ファミリー (TNF receptor family) のデスレセプターとの結合によって開始される。
- その他の2つの異常な細胞死経路は、ネクロプトーシス (necroptosis) (壊死とアポトーシスの特徴を持ち、特定のシグナル経路によって制御される) とパイロプトーシス (pyroptosis) であり、炎症性サイトカインの放出を引き起こし、アポトーシスを開始する可能性がある。
- オートファジー (Autophagy) は、細胞が自身の小器官を消化し、それらを再利用してエネルギーや基質を供給する栄養不足に対する適応です。このプロセスが対処できないほどのストレスがある場合、アポトーシスによる細胞死が引き起こされます。
細胞傷害および細胞死のメカニズム (Mechanisms of Cell Injury and Death)
個別の細胞傷害と死のメカニズムを議論する前に、いくつかの一般的な原則を強調する必要があります。
- 細胞が有害刺激に対する反応は、損傷の種類、持続時間、重症度に依存します。低濃度の毒素や短時間の虚血は可逆的な細胞損傷を引き起こす可能性がありますが、より高濃度の毒素や長時間の虚血は不可逆的な損傷と細胞死をもたらすことがあります。
- 有害刺激の結果は、損傷を受けた細胞の種類、状態、適応能力、遺伝的構成にも依存します。同じ損傷であっても、細胞の種類によって結果は大きく異なります。たとえば、脚の横紋筋は2〜3時間の完全な虚血にも耐えますが、心筋は20〜30分の虚血で死にます。栄養 (またはホルモン) の状態も重要であり、たとえばグリコーゲンを豊富に持つ肝細胞は、グルコース分子をすべて燃焼させた細胞よりも虚血に耐えられます。
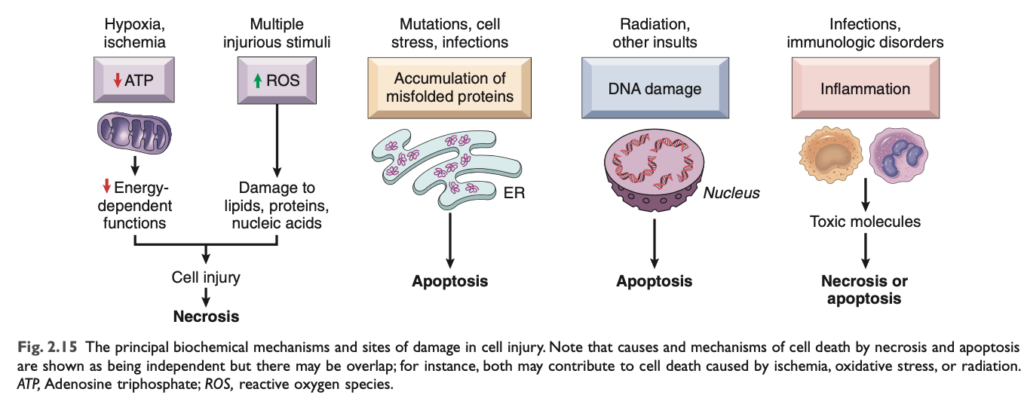
低酸素症と虚血 (Hypoxia and Ischemia)
酸素不足は、多くのエネルギー依存性代謝経路の機能不全を引き起こし、最終的には壊死による細胞死を引き起こします。ほとんどの細胞のATP は、ミトコンドリアの電子伝達系で酸素を還元する際の酸化的リン酸化 (oxidative phosphorylation) によってアデノシン二リン酸 (ADP) から生成されます。ATP は膜輸送、タンパク質合成、脂肪生成、およびリン脂質 (phospholipids) の代謝に必要な脱アシル・再アシル反応に必要です。健康な人の細胞は、毎日50〜75kgのATPを消費すると推定されています。そのため、酸素を奪われた細胞は、多くの重要な機能が壊滅的な失敗を起こすリスクが高くなります。酸素欠乏は臨床医学において細胞傷害や壊死の最も頻繁な原因の1つです。
低酸素症にさらされても即座に死なない細胞は、低酸素誘導因子1 (HIF-1) ファミリーの転写因子によって誘導される代償メカニズムを活性化します。HIF-1 は低酸素状態での細胞の生存を助けるいくつかのタンパク質の合成を促進します。これらのタンパク質の一部は、血管内皮増殖因子 (VEGF) などであり、新しい血管の成長を刺激し、血流および酸素供給の増加を試みます。HIF-1 によって誘導される他のタンパク質は、細胞代謝の適応的変化を引き起こし、グルコースの取り込み と解糖 を促進し、ミトコンドリアの酸化的リン酸化を抑制します。嫌気性解糖は、循環系または細胞内グリコーゲンの加水分解から得られるグルコースを使用して酸素なしでATPを生成できます。そのため、グリコーゲンを多く含む正常組織(例:肝臓や横紋筋)は、酸素欠乏や酸化的リン酸化の低下に耐える可能性が高いですが、グルコースの蓄えが限られている組織(例:脳)はそうではありません。速く増殖する正常細胞やがん細胞がエネルギーの多くを好気性解糖 に頼るという現象は、ワーバーグ効果 (Warburg effect) として知られています。解糖は酸化的リン酸化よりも1分子のグルコースから得られるATP量が少ないものの、解糖とTCA回路 で生成される代謝産物は細胞の成長や分裂に必要なタンパク質、脂質、核酸 などの細胞成分の合成の前駆体として役立つためです。細胞代謝の変化はがん細胞で頻繁に見られます。このため、がん細胞における詳細についてはChapter 6で議論されます。
持続的または重度の低酸素症および虚血は最終的にATP生成の失敗を引き起こし、細胞内でのATP枯渇につながります。この重要なエネルギー供給源の喪失は、細胞の多くのシステムに有害な影響を与えます (図2.16)。
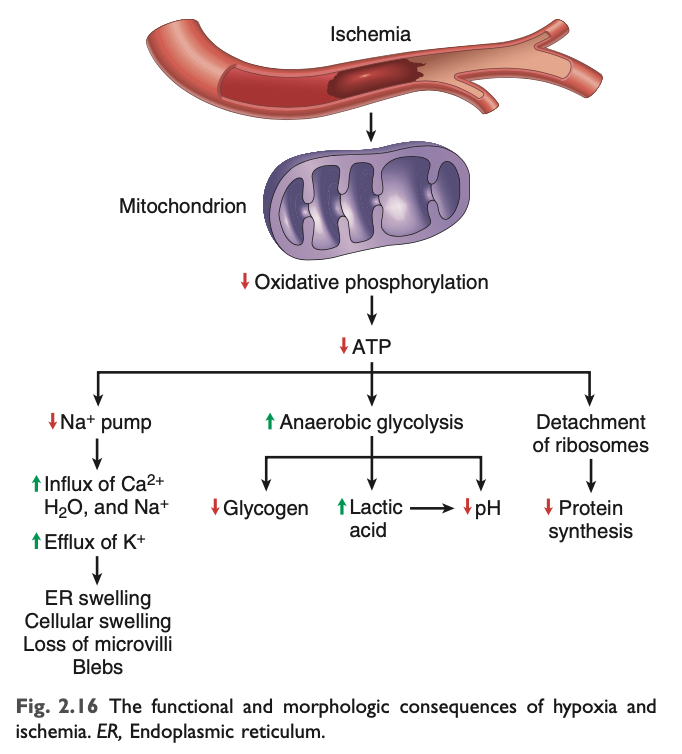
- ATP依存性ナトリウムポンプ (sodium pumps) の活性低下により、細胞内にナトリウムが蓄積し、カリウムが流出します。浸透圧に伴う水の流入が起こり、細胞の膨張や小胞体の拡張を引き起こします。
- 嫌気性解糖 の代償的な増加は、乳酸の蓄積 を引き起こし、細胞内pH の低下および多くの細胞酵素の活性低下につながります。
- ATP枯渇 が長引くと、タンパク質合成装置の構造的破壊が引き起こされ、粗面小胞体 (RER) からのリボソームの脱離やポリソームのモノソームへの解離が起こり、タンパク質合成の減少が見られます。
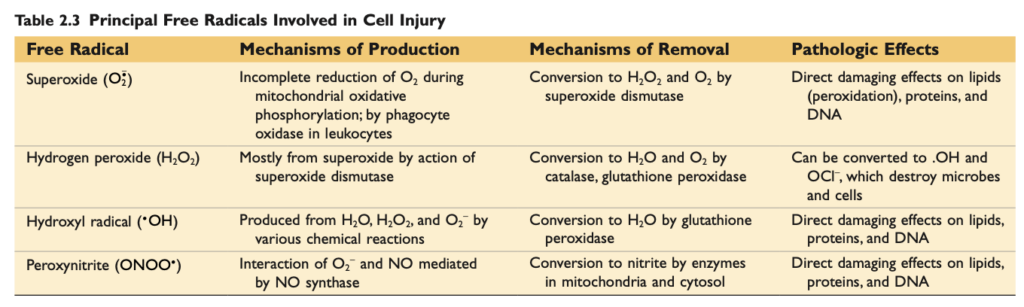
- 低酸素そのものが活性酸素種 (ROS) の蓄積を増加させる可能性も示唆されていますが、これについては議論があります。しかし、低酸素は血流と酸素供給が再開されるとROS による損傷を引き起こしやすくするという証拠が多く、この現象は再灌流傷害 (reperfusion injury) と呼ばれます。
- 最終的には、ミトコンドリア やリソソーム膜 に不可逆的な損傷が生じ、細胞は壊死に至ります。膜の損傷は、さまざまなメカニズムによる細胞傷害の最終段階であり、後述します。低酸素症によって引き起こされる主要な細胞死は壊死ですが、ミトコンドリア経路によるアポトーシスも寄与していると考えられています。
低酸素症と虚血の機能的結果は、欠乏の重症度 と期間 に依存します。たとえば、心筋は冠動脈閉塞後60秒以内に収縮を停止します。低酸素状態が続くと、ATP枯渇が進行し、図2.3で示されている一連の変化がさらに進行します。
虚血再灌流傷害 (Ischemia-Reperfusion Injury)
特定の状況下では、虚血状態にあるが生存可能な組織への血流の回復が、逆説的に細胞傷害を増加させます。これは、通常、血流の回復によって可逆的損傷を受けた細胞が回復するという予想される結果とは逆の現象です。この「虚血再灌流傷害」は、特に心筋や脳の虚血後における組織損傷に大きく寄与する臨床的に重要なプロセスです。
虚血組織への再灌流によって細胞傷害が悪化する原因となるメカニズムはいくつかあります。
- 再酸素化 中に、ROS の生成が増加することで新たな損傷が引き起こされる可能性があります。損傷したミトコンドリアは酸素の完全な還元を行えず、同時に細胞の抗酸化防御機構も虚血によって損なわれるため、状況が悪化します。浸潤する白血球 によって生成されたROS も損傷を増加させる可能性があります。
- 虚血傷害によって誘発される炎症 が再灌流によって増加し、白血球や血漿タンパク質の流入が促進されます。活性化された白血球の生成物が追加の組織傷害を引き起こす可能性があります。また、補体系 (complement system) の活性化も虚血再灌流傷害に寄与する可能性があります。補体タンパク質は損傷組織または組織に沈着した抗体に結合し、その後の補体活性化によって細胞傷害および炎症が悪化します。
毒素による細胞傷害 (Cell Injury Caused by Toxins)
環境化学物質や感染性病原体によって産生される物質を含む毒素は、主に壊死による細胞死に至る細胞傷害を引き起こします。毒素による細胞傷害には、以下の2つの一般的なメカニズムがあります。
- 直接作用する毒素 (Direct-acting toxins):
一部の毒素は、細胞の重要な分子成分や細胞小器官と直接結合して作用します。たとえば、塩化水銀 (mercuric chloride) 中毒 (汚染されたシーフードの摂取によることがあります) では、水銀がさまざまな細胞膜タンパク質のスルフヒドリル基 に結合し、ATP依存性輸送 の阻害と膜透過性の増加を引き起こします。多くの抗腫瘍化学療法薬も直接的な細胞毒性効果によって細胞損傷を引き起こします。このクラスには、微生物が産生する毒素 も含まれ (Chapter 9)、これらはしばしば、タンパク質合成 やイオン輸送 など、宿主細胞の重要な機能に必要な分子を標的にして損傷を引き起こします。 - 潜在的な毒素 (Latent toxins):
多くの毒性化学物質は、最初は活性を持たず、まず活性代謝物 に変換されてから標的細胞に作用します。理解できるように、これらの毒素は通常、それらが活性化される細胞に影響を与えます。このプロセスは、通常、肝臓や他の臓器の滑面小胞体 (smooth ER) にあるシトクロムP-450 によって行われます。代謝物は、タンパク質や脂質に共有結合 することで膜損傷や細胞傷害を引き起こす可能性がありますが、最も重要な細胞傷害のメカニズムは、フリーラジカル (free radicals) の形成に関与しています。たとえば、四塩化炭素 (CCl4)(かつてはドライクリーニング業界で広く使用されていましたが、現在は禁止されています)や鎮痛薬のアセトアミノフェン (acetaminophen) はこのカテゴリに含まれます。CCl4 は主に肝臓で有毒なフリーラジカル に変換され、このフリーラジカルが細胞損傷を引き起こし、特に膜のリン脂質過酸化 (phospholipid peroxidation) によって作用します。CCl4への曝露後30分以内に、肝細胞の小胞体膜に十分な損傷が生じ、酵素 および血漿タンパク質 の合成が減少します。2時間以内に、滑面小胞体の膨張と粗面小胞体からのリボソームの脱離が発生します。また、トリグリセリド と複合体を形成してその分泌を促進するアポタンパク質 の合成が減少し、この欠陥によって肝細胞や他の細胞に脂肪 が蓄積し、CCl4 中毒の「脂肪肝」が引き起こされます。次に、ミトコンドリアの損傷 が続き、ATPストア が減少し、イオン輸送 の欠陥と細胞の膨張が進行します。最終的に、脂質過酸化によって生成された脂肪アルデヒド によって細胞膜 がさらに損傷し、細胞死に至ります。
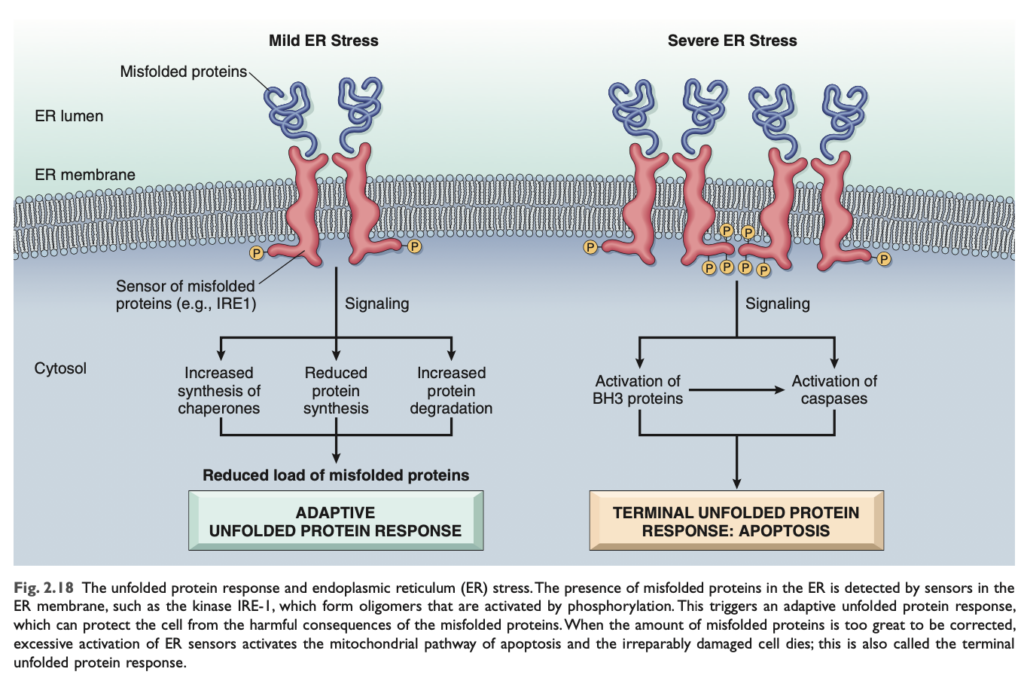
小胞体ストレス (Endoplasmic Reticulum Stress)
細胞内に誤って折り畳まれたタンパク質が蓄積すると、小胞体の代償メカニズムにストレスがかかり、最終的にアポトーシスによる細胞死が引き起こされることがあります。正常なタンパク質合成 では、小胞体内のシャペロン が新たに合成されたタンパク質の適切な折り畳みを制御し、誤って折り畳まれたポリペプチドはユビキチン化 され、プロテアソーム による分解の標的となります。小胞体に未折り畳みタンパク質 や誤って折り畳まれたタンパク質 が蓄積すると、最初に「未折り畳みタンパク質応答 (unfolded protein response)」と呼ばれる保護的な細胞応答が誘導されます (図2.18)。この適応応答は、シャペロンの産生を増加させ、タンパク質の翻訳を減少させるシグナル伝達経路 を活性化し、細胞内の誤って折り畳まれたタンパク質のレベルを低下させます。しかし、大量の誤ったタンパク質が蓄積し、適応応答では処理できなくなると、BH3ファミリー のプロアポトーシスセンサーやカスパーゼ (caspases) が活性化され、ミトコンドリア経路によるアポトーシスが引き起こされます。
DNA損傷 (DNA Damage)
細胞が放射線や化学療法剤に曝露されると、活性酸素種 (ROS) の生成や突然変異 の獲得によってDNA損傷が引き起こされることがあります。損傷が深刻な場合、アポトーシスによる細胞死を引き起こします。DNA損傷は細胞内の監視タンパク質 によって検出され、p53タンパク質 の蓄積を促進するシグナルが伝達されます。p53はまず細胞周期をG1期で停止させ、DNAが複製される前に修復される時間を与えます (Chapter 6)。しかし、損傷が大きすぎて修復が不可能な場合、p53はBH3ファミリー のセンサータンパク質を刺激し、最終的にBax とBak(Bcl-2ファミリー のプロアポトーシスメンバー)を活性化し、アポトーシスを引き起こします。p53が変異していたり欠損している場合 (特定のがんで見られるように)、損傷したDNAを持つ細胞がアポトーシスを回避して生存します。このような細胞では、DNA損傷が突然変異 や染色体再編成 (例: 転座) を引き起こし、腫瘍形成 に至ることがあります。
炎症 (Inflammation)
細胞や組織に損傷を与える一般的な原因は、病原体、壊死した細胞、または自己免疫疾患やアレルギーにおける免疫応答の異常 によって引き起こされる炎症反応 です。これらの状況すべてにおいて、好中球、マクロファージ、リンパ球 などの炎症細胞は微生物を破壊するために進化した生成物を分泌しますが、それらが宿主組織にも損傷を与える可能性があります。これらの有害な免疫反応は、過敏症 (hypersensitivity) に分類されます。
多様な原因による細胞傷害の共通のイベント (Common Events in Cell Injury From Diverse Causes)
前の議論では、細胞傷害のメカニズムを原因に応じて説明し、さまざまな病理生理学的状況で引き起こされる主要な損傷経路を強調しました。いくつかの異常は、原因にかかわらず細胞傷害に共通して見られるものであり、さまざまな病理的状況で観察されます。ここでは、そのうちの2つの変化について説明します。
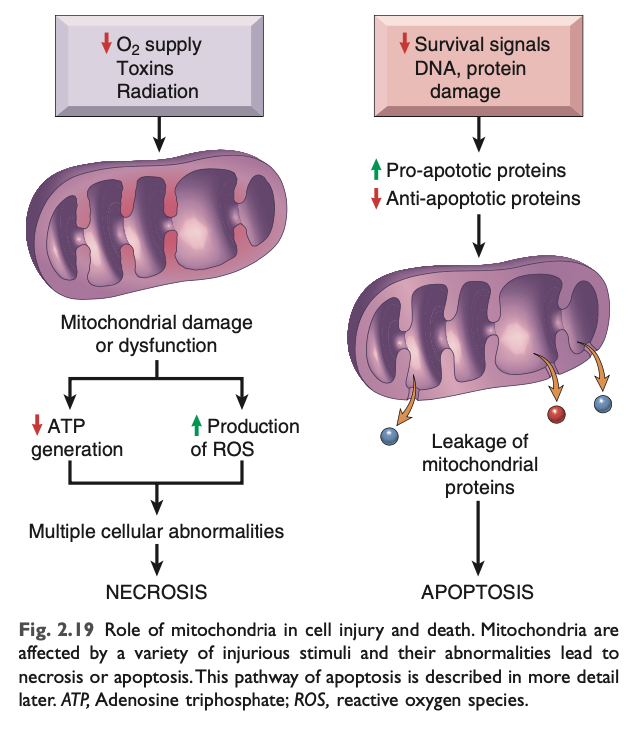
ミトコンドリア機能障害 (Mitochondrial Dysfunction)
ミトコンドリア は、ATPの形で生命を維持するエネルギーを生産する「ミニ工場」と見なすことができます。そのため、細胞傷害や細胞死においても重要な役割を果たします。ミトコンドリアは、低酸素、化学毒素、放射線 などの多くの有害刺激に敏感です (図2.19)。ミトコンドリアの変化は、壊死およびアポトーシスの両方で発生し、いくつかの生化学的異常を引き起こします。
- 酸化的リン酸化の失敗 は、ATPの枯渇を引き起こし、最終的には細胞の壊死に至ります。
- 異常な酸化的リン酸化は活性酸素種 (ROS) の生成も引き起こし、さまざまな有害な影響を与えます。
- ミトコンドリア膜 に損傷が生じると、ミトコンドリア透過性転移孔 (mitochondrial permeability transition pore) と呼ばれる高伝導チャネルが形成されます。このチャネルが開くと、ミトコンドリア膜電位が失われ、pH変化が生じ、酸化的リン酸化がさらに損なわれます。
- ミトコンドリアには、シトクロムC (cytochrome c) のようなタンパク質が含まれており、これが細胞質に放出されると、細胞内の損傷を知らせ、アポトーシス経路を活性化します。
膜透過性の欠陥 (Defects in Membrane Permeability)
膜の透過性の増加が最終的に顕著な膜損傷に至ることは、壊死に至るほとんどの細胞傷害に共通する特徴です。細胞傷害中に最も重要な膜損傷の部位は、ミトコンドリア膜、細胞膜、およびリソソーム膜 です。細胞膜およびリソソーム膜の透過性の増加は、アポトーシスでは見られません。
- ミトコンドリア膜の損傷: ミトコンドリア膜の損傷は、ATPの産生を減少させ、最終的には壊死に至る多くの有害な影響を引き起こします。
- 細胞膜の損傷: 細胞膜の損傷は、浸透圧バランスの喪失を引き起こし、体液やイオンの流入、さらには細胞内容物の喪失につながります。細胞は、ATP の再合成に重要な代謝産物を漏出することもあり、エネルギー貯蔵がさらに枯渇します。
- リソソーム膜の損傷: リソソーム膜の損傷は、酵素が細胞質に漏れ出し、損傷した細胞内の酸性pHで酸性加水分解酵素が活性化されます。これにより細胞成分の酵素的消化が引き起こされ、細胞は壊死に至ります。
要約 (SUMMARY)
細胞傷害のメカニズム (Mechanisms of Cell Injury)
- さまざまな開始イベントは、異なるメカニズムを介して細胞傷害および細胞死を引き起こします。
- 低酸素症 および虚血 は、ATPの枯渇を引き起こし、多くのエネルギー依存的機能の失敗をもたらします。これにより、最初は可逆的な損傷が発生し、修復されない場合は壊死に至ります。
- 虚血再灌流傷害 (ischemia-reperfusion injury) では、虚血組織への血流の回復がROS の生成や炎症を増加させ、損傷を悪化させます。
- 酸化ストレス (oxidative stress) は、ROS の蓄積を指し、細胞の脂質、タンパク質、およびDNA に損傷を与え、さまざまな原因に関連しています。
- タンパク質の誤折り畳み (protein misfolding) は、必須タンパク質の枯渇を引き起こし、誤折り畳みタンパク質が細胞内に蓄積すると、アポトーシスが発生します。
- DNA損傷 (例:放射線による) も修復されない場合、アポトーシスを引き起こします。
- 炎症 (inflammation) は、炎症性白血球の生成物による損傷作用のため、細胞傷害と関連しています。
ストレスに対する細胞の適応 (Cellular Adaptations to Stress)
適応とは、環境の変化に応じて、細胞の数、サイズ、表現型、代謝活動、または機能が可逆的に変化することです。生理的適応 (Physiologic adaptations) は通常、ホルモンや内因性化学物質による正常な刺激 (例:妊娠中の乳房や子宮の拡大) や、機械的ストレスの要求 (例:骨や筋肉の場合) に対する細胞の反応を示します。一方、病理的適応 (Pathologic adaptations) は、細胞が損傷を逃れるために構造や機能を調節する反応であり、正常な機能を犠牲にすることがあります (例:喫煙者の気管支上皮の扁平上皮化生 (squamous metaplasia))。生理的および病理的な適応には、いくつかの異なる形態があります。
肥大 (Hypertrophy)
肥大 は細胞サイズの増加による臓器サイズの増加です。対照的に、過形成 (hyperplasia) は細胞数の増加です。言い換えれば、純粋な肥大では新しい細胞はなく、構造タンパク質や小器官の量が増加しただけの大きな細胞が存在します。過形成 は分裂可能な細胞の適応反応である一方で、肥大は分裂能力が限られた細胞で起こります。肥大と過形成は同時に発生することがあり、両方とも臓器の拡大を引き起こします。
肥大には生理的肥大 と病理的肥大 があり、これは機能的需要の増加や成長因子 やホルモン の刺激によって引き起こされます。
- 生理的肥大の例: 妊娠中にエストロゲン 刺激によって平滑筋肥大 および過形成 が起こり、子宮が大きくなります。一方、骨格筋 や心筋 のような横紋筋細胞は、分裂能力が限られている ため、負荷の増加に応じて肥大のみが起こります。
- 病理的肥大の例: 高血圧や大動脈弁疾患 による心筋の肥大が挙げられます。心筋が負荷の増加に適応するために肥大を引き起こし、収縮力を高めますが、過度のストレスが続くと肥大は不可逆的な損傷に進行する可能性があります。
過形成 (Hyperplasia)
過形成は、分化した細胞または未分化な前駆細胞の増殖による臓器内の細胞数の増加を指します。過形成は、分裂可能な細胞 が存在する組織で発生し、しばしば肥大と同時に発生します。
- 生理的過形成の例: 1) ホルモン性過形成 は、思春期や妊娠中に見られる乳腺上皮 の増殖を示し、2) 代償性過形成 は、臓器の一部が切除されたり失われた後に残存組織が増殖して臓器を再生するものです。
- 病理的過形成の例: 過剰なホルモン または成長因子 の刺激によって引き起こされるもので、たとえば子宮内膜過形成 や前立腺肥大 が挙げられます。
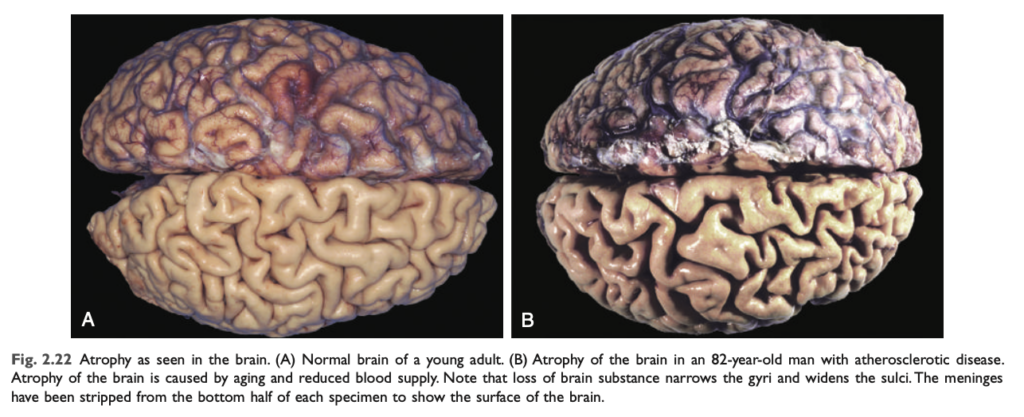
萎縮 (Atrophy)
萎縮は、細胞物質の減少 による細胞サイズの縮小です。多くの細胞が関与する場合、組織や臓器全体が縮小し、萎縮します。萎縮した細胞は機能が低下 しますが、死んでいるわけではありません。
萎縮の原因には、負荷の減少 (例:骨折の治癒を促進するための四肢の固定)、神経支配の喪失、血流の減少、栄養不足、内分泌刺激の喪失 などがあります。
化生 (Metaplasia)
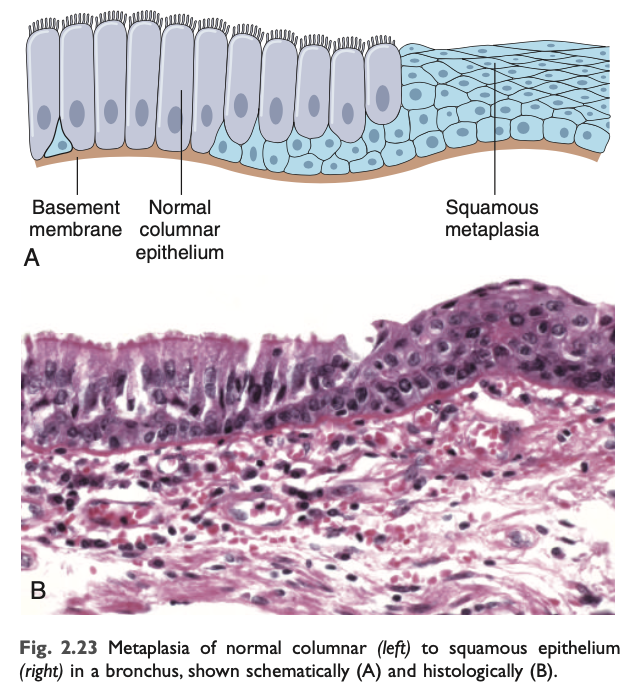
化生は、一つの成熟細胞型 (上皮または間葉系) が、別の成熟細胞型に置き換えられる変化です。これは、特定のストレスに敏感な細胞が、より耐性のある細胞型 に置き換わる適応です。たとえば、喫煙者の気道上皮では、正常な線毛円柱上皮細胞 が重層扁平上皮 に置き換えられます。
上皮化生 (epithelial metaplasia) の例として、習慣的な喫煙者の気道上皮 に見られる変化があります。喫煙者の気管 や気管支 では、通常の線毛円柱上皮細胞 が重層扁平上皮細胞 に置き換えられます (図2.23)。この丈夫な重層扁平上皮 は、より脆弱な専門化した上皮細胞 よりも、タバコの煙に含まれる有害な化学物質に対して耐性があります。しかし、化生した扁平上皮には生存上の利点 がある一方で、粘液分泌 や繊毛による異物の除去 といった重要な防御機能が失われるため、化生は両刃の剣 です。
ビタミンA は正常な上皮の分化に必要であるため、その欠乏は呼吸器上皮における扁平上皮化生 を引き起こすこともあります。化生は、円柱上皮から扁平上皮 への変化だけでなく、逆に起こることもあります。たとえば、慢性的な胃食道逆流症 (GERD) の場合、下部食道 の正常な重層扁平上皮 は、胃 や腸 型の円柱上皮 に化生することがあります。化生は間葉系細胞 にも起こり得ますが、これらの状況では、病理的な変化に対する反応であり、ストレスに対する適応ではありません。たとえば、軟部組織 に骨 が形成されることがあり、特に損傷部位で発生します。
上皮に化生変化を引き起こす影響が持続する場合、これが悪性変換 の素因となる可能性があります。事実、呼吸器上皮の扁平上皮化生 は、しばしば悪性扁平上皮細胞 で構成された肺癌 と共存します。喫煙は最初に扁平上皮化生 を引き起こし、後にこれらの変化した部位の一部に癌 が発生することがあると考えられています。
ストレスに対する細胞の適応 (Summary: Cellular Adaptations to Stress)
- 肥大 (Hypertrophy): 細胞や臓器のサイズの増加。通常は機能的負荷の増加に応じて発生し、機械的ストレスや他の刺激に応じて産生される成長因子によって誘導される。細胞分裂ができない組織で発生する。
- 過形成 (Hyperplasia): 細胞数の増加。ホルモンや他の成長因子に応じて発生し、細胞分裂が可能な組織や豊富な組織幹細胞を持つ組織で発生する。
- 萎縮 (Atrophy): 栄養供給の減少や不使用による細胞や臓器サイズの減少。細胞の構成成分の合成が減少し、細胞小器官やオートファジーの分解が増加することが関係している。
- 化生 (Metaplasia): 分化した細胞の表現型の変化。しばしば慢性的な刺激に応じて発生し、細胞がストレスに耐えやすくなるようにする。通常は、組織幹細胞の分化経路の変化によって誘導され、機能が低下するか、または悪性変換の可能性が増加することがある。
細胞内蓄積物 (Intracellular Accumulations)
特定の状況下で、細胞はさまざまな物質を異常に蓄積することがあり、それは無害であるか、ある程度の損傷を引き起こすことがあります。これらの物質は、細胞質、小器官 (通常はリソソーム) 内、または核内に存在し、細胞自体によって合成されるか、別の場所で産生されたものが蓄積します。
異常な細胞内蓄積の主な経路は、内因性物質の除去および分解の不十分さ、または過剰な産生、あるいは異常な外因性物質の沈着です。以下にそれぞれの例を挙げます。
- 脂肪変性 (Fatty Change): 別名ステアトーシスは、実質細胞内にトリグリセリド が蓄積することを指します。これは主に脂肪代謝に関与する肝臓 に見られますが、心臓、骨格筋、腎臓、他の臓器でも発生することがあります。原因には、毒素、タンパク質栄養不良、糖尿病、肥満、無酸素症などが含まれます。アルコール乱用 や肥満に関連する糖尿病は、工業化された国々で最も一般的な脂肪変性 (脂肪肝) の原因です。
- コレステロールおよびコレステリルエステル (Cholesterol and Cholesteryl Esters): コレステロール は細胞膜の重要な構成要素であり、その代謝は厳密に調整されています。しかし、病理過程において、脂質 (トリグリセリド、コレステロール、コレステリルエステル) がファゴサイト に過剰に蓄積することがあります。この中で、動脈硬化症 が最も重要です。
- タンパク質 (Proteins): タンパク質の異常蓄積 は、過剰な量が細胞に提示されたり、細胞が過剰な量を合成した場合に発生します。たとえば、腎臓では、通常、微量のアルブミン が糸球体を通過して近位尿細管で再吸収されますが、腎症候群 のように、タンパク質の漏出が多い場合、再吸収されたタンパク質が蓄積し、ヒアリン状の細胞質の小滴 が形成されます。この過程は可逆的で、タンパク尿が治まると、これらの小滴は代謝されて消失します。
- グリコーゲン (Glycogen): グリコーゲンの異常な蓄積は、グルコース またはグリコーゲンの代謝異常に関連しています。糖尿病 では、グリコーゲンが腎尿細管上皮細胞、心筋細胞、ランゲルハンス島のβ細胞 に蓄積します。グリコーゲン蓄積が見られる遺伝性疾患は、グリコーゲン貯蔵病 として知られています。
- 色素 (Pigments): 色素は、外因性 (体外から取り込まれる) または内因性 (体内で合成される) の色素であり、たとえば、リポフスチン、メラニン、ヘモグロビン の代謝産物などがあります。
- リポフスチン (Lipofuscin): 「摩耗色素」とも呼ばれるもので、老化や萎縮に伴って心臓、肝臓、脳 に蓄積します。これは脂質 とタンパク質 の複合体であり、細胞膜のポリ不飽和脂質 のフリーラジカルによる過酸化 によって生成されます。リポフスチンは細胞に害を及ぼさないものの、過去のフリーラジカル損傷のマーカーです。
- メラニン (Melanin): メラニン は内因性の黒色の色素で、表皮に存在するメラノサイト によって合成され、紫外線 からの保護スクリーンとして機能します。
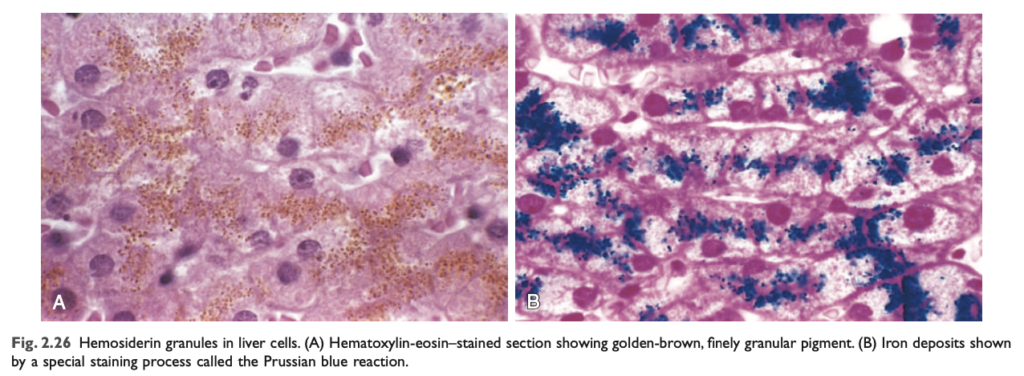
- ヘモジデリン (Hemosiderin): ヘモグロビン 由来の粒状色素であり、主に鉄 の局所または全身的な過剰がある場合に蓄積します。
- ヘモジデリンは、ヘモグロビン 由来の黄金色から茶色の粒状色素 であり、局所的または全身的に鉄 が過剰に存在する場合に組織 に蓄積します。通常、鉄は細胞内でアポフェリチン というタンパク質と結合してフェリチンミセル を形成し、これが鉄を貯蔵します。ヘモジデリン 色素は、これらのフェリチンミセルの大規模な凝集体 を表しており、光学顕微鏡および電子顕微鏡で容易に観察できます。鉄はプルシアンブルー 染色によって明確に識別されます (図2.26)。ヘモジデリンの蓄積は通常、病的 なものであり、骨髄、脾臓、肝臓 の単核ファゴサイトで少量のヘモジデリンが見られるのは正常なことですが、これらは老化した赤血球 が通常分解される場所です。
- 過剰なヘモジデリンの沈着をヘモジデローシス (hemosiderosis) と呼び、さらに進行した鉄の蓄積は、遺伝性のヘモクロマトーシス (hemochromatosis) で見られます。この状態についてはChapter 16 で詳しく説明されています。
病理学的石灰化 (Pathologic Calcification)
病理学的石灰化は、さまざまな疾患状態でよく見られる過程であり、カルシウム塩 の異常な沈着の結果であり、これには少量の鉄、マグネシウム、および他の鉱物も含まれます。これには、2つの形態があります。
- 異栄養性石灰化 (Dystrophic Calcification): このタイプでは、カルシウム代謝は正常ですが、損傷または死んだ組織にカルシウムが沈着します。例えば、壊死が発生した領域です。この石灰化は、進行した動脈硬化症 (atherosclerosis) の動脈病変でほぼ必ず見られます。異栄養性石灰化は、過去の軽度な細胞損傷を示す偶発的な発見であることもありますが、臓器機能障害の原因となることもあります。例えば、心臓弁 が老化または損傷すると、石灰化が進行し、弁の動きが著しく制限されることがあります。大動脈弁の異栄養性石灰化 は、高齢者における大動脈狭窄症 (aortic stenosis) の重要な原因です。
- 異栄養性石灰化は、損傷した細胞から由来する膜結合小胞や、死んだ細胞のミトコンドリア 内にカルシウムが沈着することによって始まります。カルシウムは膜リン脂質への親和性によって小胞内に集中し、膜結合ホスファターゼ の働きによりリン酸 が蓄積すると考えられています。その結果、結晶が形成され、沈着物が増大します。
- 転移性石灰化 (Metastatic Calcification): このタイプは、高カルシウム血症 (hypercalcemia) に関連しており、正常組織に発生します。高カルシウム血症の主な原因には、以下が含まれます。
- 副甲状腺ホルモン (PTH) の分泌増加 (副甲状腺腫瘍または他の悪性腫瘍によるPTH関連タンパク質の産生)
- 骨の破壊 (例:パジェット病 (Paget disease))、骨髄腫、白血病、または骨転移 に伴う骨の異常な崩壊
- ビタミンD関連障害 (例:ビタミンD中毒、サルコイドーシス (sarcoidosis) など、マクロファージがビタミンD前駆体を活性化)
- 腎不全 (renal failure) によるリン酸の蓄積 とそれに伴う二次性副甲状腺機能亢進症。
形態学 (Morphology)
カルシウム塩は、肉眼 で白色の細かい粒や塊として見られ、しばしばザラザラした沈着物として感じられます。異栄養性石灰化は、結核 (tuberculosis) における乾酪壊死 (caseous necrosis) の領域で一般的に見られます。時には、結核性リンパ節が放射線不透過性の石 に変わることもあります。組織学的検査 では、石灰化は細胞内 または細胞外 の好塩基性沈着物 として現れます。時間が経つと、石灰化の部位で異所性骨形成が起こることがあります。
転移性石灰化は全身に広く分布する可能性がありますが、特に血管間質、腎臓、肺、および胃粘膜 に影響を与えます。カルシウム沈着の形態学的特徴は、異栄養性石灰化と類似しています。一般的には臨床的な機能障害を引き起こしませんが、肺での広範な石灰化はX線画像 によって明らかになり、呼吸機能の低下を引き起こすことがあります。また、腎臓における大量の石灰沈着 (腎石灰症 (nephrocalcinosis)) は、腎障害 を引き起こす可能性があります。
要約 (Summary)
異常な細胞内沈着物および石灰化 (Abnormal Intracellular Depositions and Calcifications)
- 異常な物質の沈着 は、過剰な摂取 または輸送や代謝異常 の結果として細胞や組織に発生します。
- 脂質の沈着
- 脂肪変性 (Fatty Change): トリグリセリド の細胞内蓄積。過剰摂取または輸送異常 (特に輸送タンパク質の合成異常) によって引き起こされる。可逆的な細胞損傷 の兆候。
- コレステロール沈着: 代謝異常 や過剰摂取 の結果として、動脈硬化症 の血管壁のマクロファージや平滑筋細胞に見られる。
- タンパク質の沈着: 腎尿細管で再吸収されたタンパク質や、形質細胞 の免疫グロブリン など。
- グリコーゲンの沈着: リソソーム酵素異常 によるグリコーゲンの分解異常 (グリコーゲン貯蔵病) に関連する。
- 色素の沈着: 炭素 やリポフスチン (脂質過酸化の分解生成物)、鉄 (ヘモジデローシスに関連する) などの、通常消化されない色素。
- 病理学的石灰化
- 異栄養性石灰化: 細胞損傷や壊死 の部位へのカルシウム沈着。
- 転移性石灰化: 高カルシウム血症 による正常組織へのカルシウム沈着。通常、副甲状腺ホルモン過剰に関連。
細胞の老化 (Cellular Aging)
個人が老化する理由は、その細胞が老化 するためです。従来、老化プロセスに対する関心は美容的な側面 に焦点を当ててきましたが、老化には重要な健康への影響があり、がん、アルツハイマー病、虚血性心疾患 など、多くの慢性疾患の独立した強力なリスク要因 の一つです。細胞老化に関する最も驚くべき発見の一つは、単に細胞が「エネルギーを使い果たす」結果ではなく、限られた遺伝子 やシグナル伝達経路 によって調節されており、これらは酵母から哺乳類に至るまで進化的に保存 されているという事実です。
細胞の老化 は、細胞の寿命と機能的活動の進行性の低下によって引き起こされます。細胞老化にはいくつかの異常が関与しています (図2.27)。
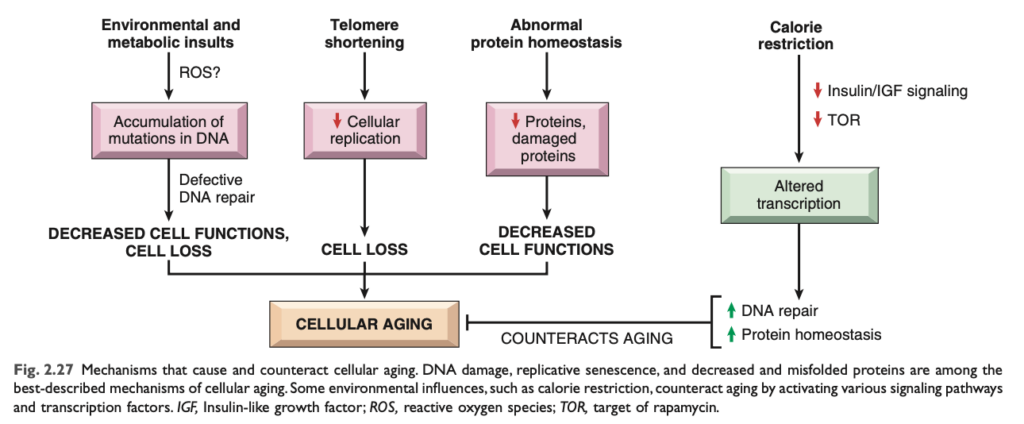
- DNAの突然変異の蓄積: 長期間にわたるさまざまな代謝的な損傷は、核やミトコンドリアのDNAに損傷を与える可能性があります。毒素 や放射線暴露 によって引き起こされる活性酸素種 (ROS) は、老化に関連するDNA損傷に寄与します。ほとんどのDNA損傷はDNA修復酵素 によって修復されますが、一部は修復されず、細胞が老化するにつれて蓄積します。最終的に、核およびミトコンドリアのDNAの突然変異の蓄積は、細胞の機能的活動や生存に影響を及ぼします。
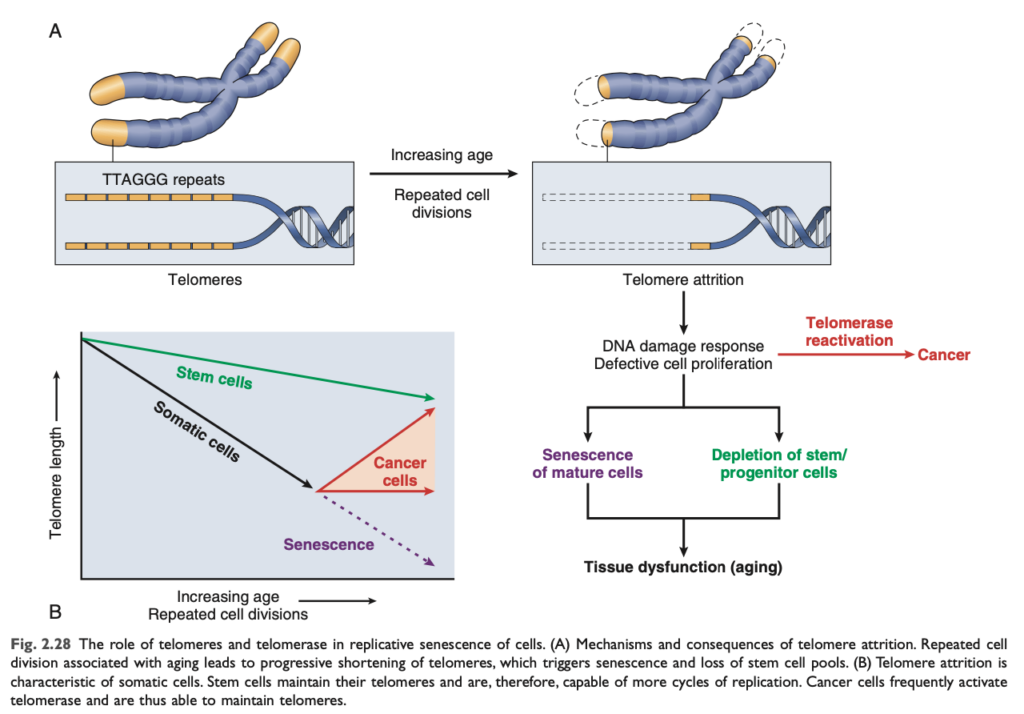
- 細胞複製の減少: 正常な細胞 (幹細胞を除く) は複製能力が限られて おり、一定の回数の分裂の後、複製老化 (replicative senescence) と呼ばれる終末的な非分裂状態に入ります。複製老化は、テロメア の進行性の短縮によって引き起こされ、最終的には細胞周期停止 につながります。テロメアは、染色体の末端に存在する短い繰り返し配列のDNA であり、染色体の完全な複製と末端保護 に重要です。細胞が複製するたびにテロメアは短縮し、染色体末端が壊れたDNA として認識されると、細胞周期が停止します。テロメラーゼ (telomerase) という酵素が、テロメアの長さを維持する役割を果たしますが、この酵素は生殖細胞 や低レベルで幹細胞に存在するものの、体細胞 にはほとんど存在しません。老化した体細胞ではテロメアが短縮し、新しい細胞を生成できなくなります。一方で、がん細胞 ではテロメラーゼが再活性化され、無限に増殖する能力を持つことがあります。
- タンパク質恒常性の欠陥: 時間の経過とともに、細胞は正常なタンパク質恒常性 を維持できなくなります。これは翻訳の減少 やシャペロン (正常なタンパク質の折り畳みを促進) とプロテアソーム (誤って折り畳まれたタンパク質を分解) の活動不全によるものです。これにより、細胞内のタンパク質の減少 や誤って折り畳まれたタンパク質の蓄積 が進行し、細胞の生存や複製に悪影響を及ぼす可能性があります。
- カロリー制限による老化抑制: カロリー制限が老化を抑え、寿命を延ばすことが、多くの生物種で確認されています。これは、インスリン様成長因子 (IGF-1) シグナル伝達の活性化が減少することによるものと考えられています。IGF-1シグナルの低下は、細胞の成長や代謝を減少させ、DNA複製のエラーが減少し、DNA修復 やタンパク質恒常性 が改善されると考えられています。
- 持続的炎症: 年齢を重ねると、損傷した細胞や脂質などが蓄積し、インフラマソーム経路 を活性化させ、持続的な低レベルの炎症 が引き起こされます。この炎症が慢性疾患 (例: 動脈硬化、2型糖尿病) を引き起こし、炎症中に産生されるサイトカイン は、老化をさらに悪化させます。
要約 (Summary)
細胞の老化 (Cellular Aging)
- 細胞の老化は、以下を含む複数の進行性の細胞異常の組み合わせによって引き起こされます:
- DNA損傷および突然変異の蓄積
- 複製老化 (replicative senescence): 染色体末端 (テロメア) の進行性の短縮による細胞分裂能力の低下
- タンパク質恒常性の欠陥: 正常なタンパク質の喪失と誤った折り畳まれたタンパク質の蓄積
- 老化は、慢性疾患 (特に長期間の炎症を伴うもの) やストレス によって悪化し、カロリー制限 や運動 によって遅くなります。

コメント