
学習目標
- ミオグロビンとヘモグロビンの最も重要な構造的類似点と相違点を説明する。
- ミオグロビンとヘモグロビンの酸素化(oxygenation)に関する結合曲線を区別して描写する。
- ヘモグロビンのシグモイド(sigmoidal)曲線と、ミオグロビンの双曲線的な酸素結合曲線の違いを説明する。
- ヘモグロビンの酸素化および脱酸素化に伴う構造的および立体的変化を説明する。
- **2,3-ビスホスホグリセリン酸(BPG)**が酸素結合と供給に果たす役割を説明する。
- **ボーア効果(Bohr Effect)**が赤血球のCO2輸送を促進するメカニズムを説明する。
- **ヘモグロビン異常症(hemoglobin disorders)**を解説する。
Contents
- 0.1 今日のディスカッション
- 0.2 イントロダクションと専門用語
- 0.3 ミオグロビン (Myoglobin)
- 0.4 ヘモグロビン (Hemoglobin)
- 0.5 酸素解離曲線 (Oxygen Dissociation Curves)
- 0.6 ヘモグロビンの立体構造変化(Conformational Changes in Hemoglobin)
- 0.7 ボーア効果(Bohr Effect)とCO2輸送
- 0.8 その他の要因
- 0.9 臨床的意義(Clinical Significance)
- 0.10 ミオグロビン尿症(Myoglobinuria)
- 0.11 貧血(Anemia)
- 0.12 サラセミア(Thalassemias)と他のヘモグロビン異常症(Hemoglobinopathies)
- 0.13 一酸化炭素中毒(Carbon Monoxide Poisoning)
- 0.14 高山病(Altitude Sickness)
- 1 Proteins: Myoglobin & Hemoglobin
今日のディスカッション
トピック概要
- イントロダクションと専門用語の説明
- ミオグロビンについて
- ヘモグロビンについて
- 酸素結合曲線について
- ボーア効果とその他の要因について
- 臨床的意義
イントロダクションと専門用語
ミオグロビン (Myoglobin):
- ミオグロビンは赤筋(red muscle)の単量体タンパク質で、酸素不足に備えて酸素を強く結合します。
ヘモグロビン (Hemoglobin):
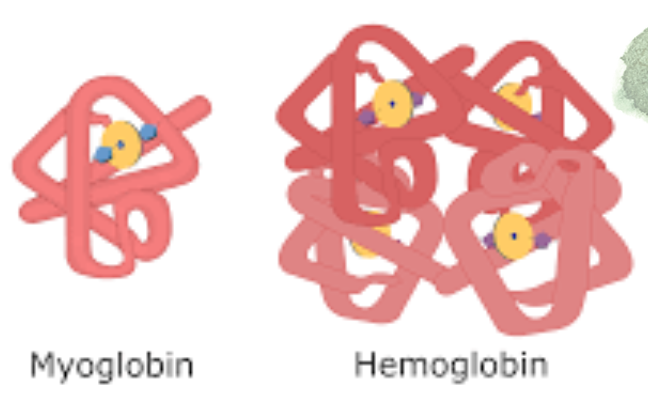
- ヘモグロビンは赤血球(erythrocytes)内の四量体タンパク質で、協同的な作用により末梢組織で酸素を高効率に放出しつつ、肺では効率的に酸素を結合します。
ヘム (Heme):
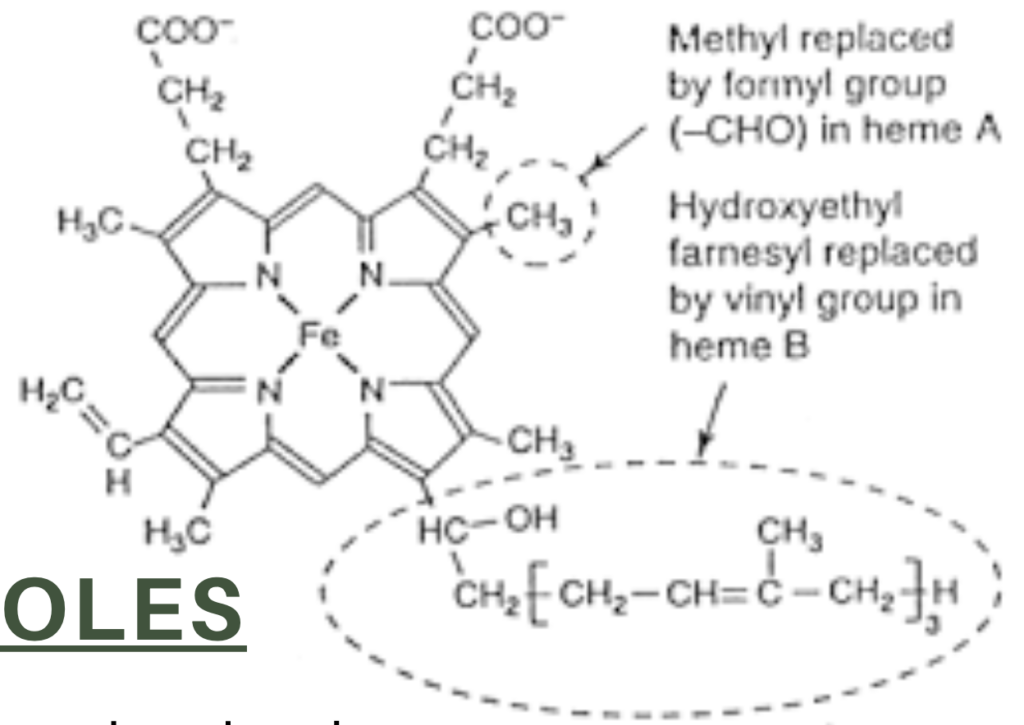
- ヘムは、四つのピロール(pyrrole)分子がメチン橋(methyne bridges)で結合した鉄含有環状テトラピロールです。

テトラピロール (Tetrapyrroles):
- テトラピロールは、四つの五員環状ヘテロ環式(pyrrole)リングが環状または線状に結合した有機分子です。
グロビン (Globins):
- グロビンは、ヘム補欠分子族(heme prosthetic group)を持つタンパク質のファミリーで、細胞内で様々な生物学的機能に関与します。
ミオグロビン (Myoglobin)
構成:
- ミオグロビンは、中心にヘム補欠分子を持つ単一鎖の球状タンパク質です。
- ヘムの中心にある鉄は、ポルフィリン環の四つの窒素原子、His-64のイミダゾール側鎖、酸素分子と結合します。
機能:
- 筋肉における酸素の貯蔵
- 酸素不足(例: 激しい運動時)で酸素を放出し、筋肉ミトコンドリアでのATP合成に使用されます。
ヘモグロビン (Hemoglobin)
構成:
- ヘモグロビンは、タンパク質成分とポルフィリン誘導体の鉄錯体からなる血液中の赤色色素です。
- 四次構造: 異なる二つのタンパク質対で構成され、α鎖(α chains)とβ鎖(β chains)に分けられます。
- 異なるサブユニット:
- HbA: 正常な成人のヘモグロビン
- HbF: 胎児のヘモグロビン
- HbS: 鎌状赤血球(sickle cell)ヘモグロビン
- HbA2: 成人のマイナーなヘモグロビン
機能:
- 肺から組織への酸素の輸送
- 末梢組織から肺への二酸化炭素の輸送
- アロステリック効果 (Allosteric Effect):
- 一つのサブユニットが酸素を結合すると、他のサブユニットが酸素をより容易に結合できるようになります。
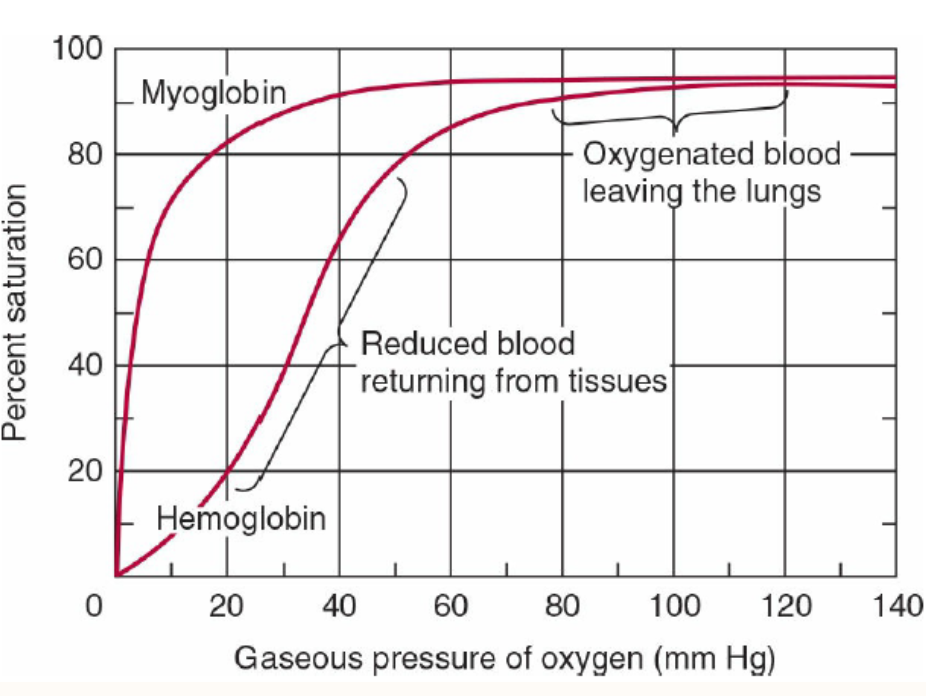
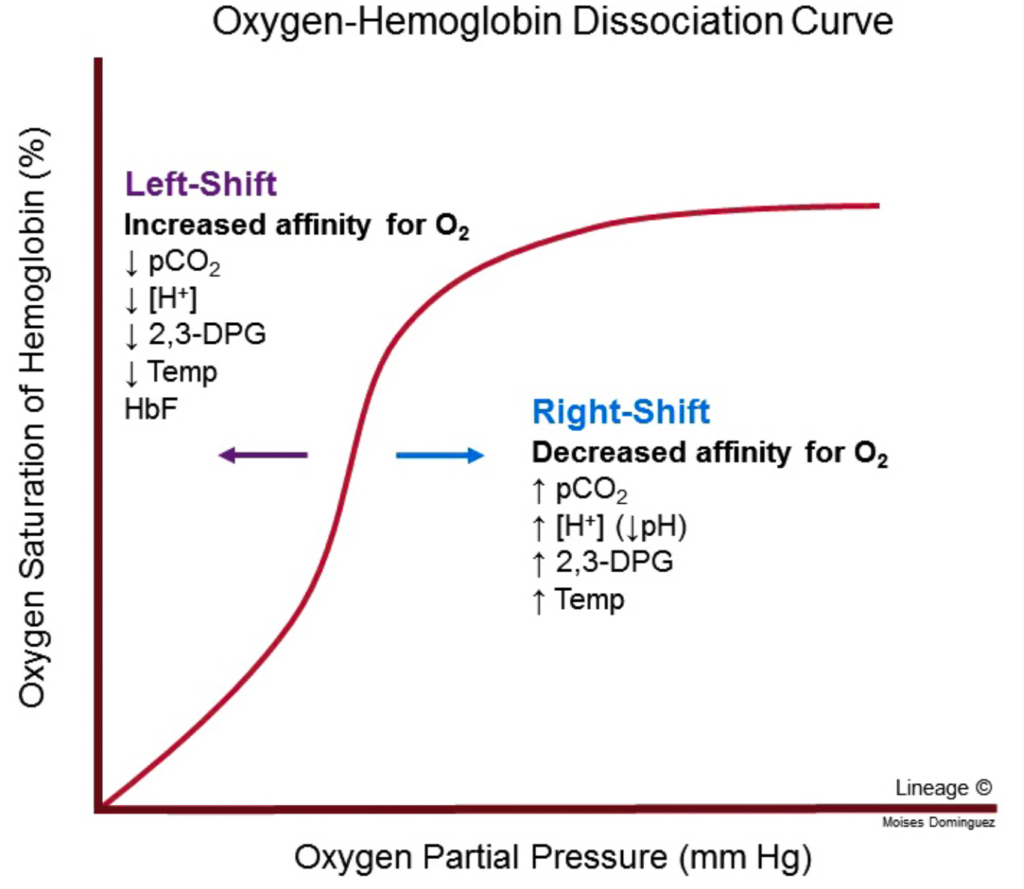
酸素解離曲線 (Oxygen Dissociation Curves)
ミオグロビン (Myoglobin):
- 双曲線的酸素結合曲線 (Hyperbolic O2-binding Curve)
- 肺の毛細血管床の酸素分圧で容易に酸素を結合しますが、活動中の筋肉や他の組織で通常見られる酸素分圧では結合した酸素のわずかな部分しか放出しません。
- 酸素運搬には効果的ではありません。
- 激しい運動により筋肉組織の酸素分圧が低下し、ミオグロビンから酸素が解離し、筋肉活動を継続するためのミトコンドリアでのATP合成が可能になります。
ヘモグロビン (Hemoglobin):
- シグモイド酸素結合曲線 (Sigmoid O2 Binding Curve)
- ヘモグロビンはあたかも二つの異なるタンパク質のように振る舞います。
- 高い酸素分圧では高い酸素親和性(R/リラックス状態)を示します(肺)。
- 低い酸素分圧では低い酸素親和性(T/タイト状態)を示します(末梢組織)。
- これにより、組織への酸素供給が可能になります。
ヘモグロビンの立体構造変化(Conformational Changes in Hemoglobin)
- 最初の酸素分子の結合: 脱酸素ヘモグロビン(deoxyHb)に最初の酸素(O2)分子が結合すると、ヘム鉄がヘム環の平面に近づきます。
- 構造の変化: 酸素誘導によってヘモグロビンが低親和性のT状態から高親和性のR状態へ移行する際、二次構造(secondary structure)、三次構造(tertiary structure)、四次構造(quaternary structure)に大きな変化が生じます。
- 親和性の増加: これにより、残りの未酸素化ヘムの酸素に対する親和性が大幅に向上し、次の酸素分子が結合する際には、塩橋(salt bridges)の破壊が少なくて済みます。

ボーア効果(Bohr Effect)とCO2輸送
- CO2の輸送: CO2は赤血球(RBC)によって回収され、肺で処理されます。静脈血の15%はカルバミン酸塩(carbamates)として運ばれます。
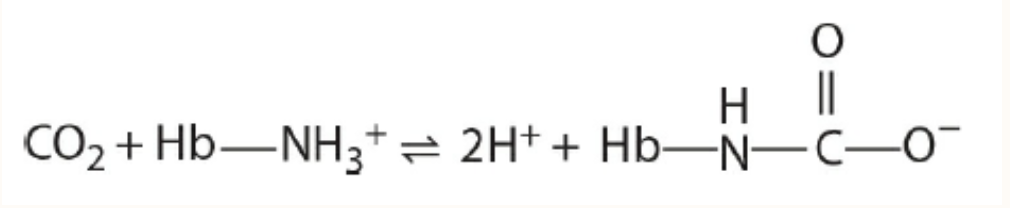
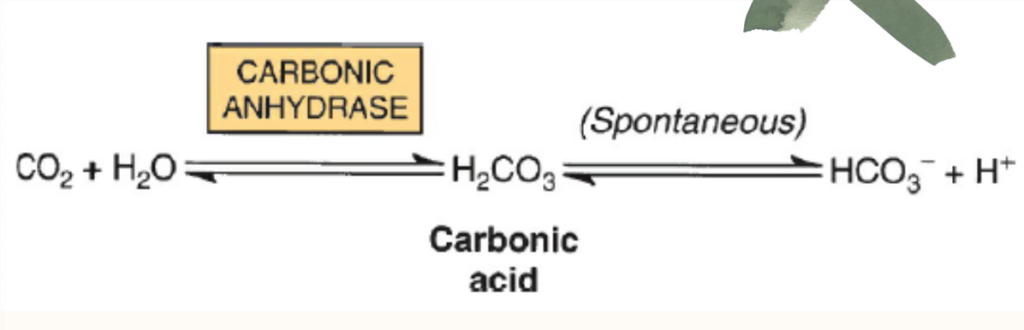
- 炭酸(Carbonic Acid): 残りのCO2は主に炭酸(H2CO3)として赤血球内でCO2の水和によって形成されます。この反応は炭酸脱水酵素(carbonic anhydrase)によって触媒されます。静脈血のpHでは、炭酸は重炭酸塩(bicarbonate)とプロトン(H+)に解離します。
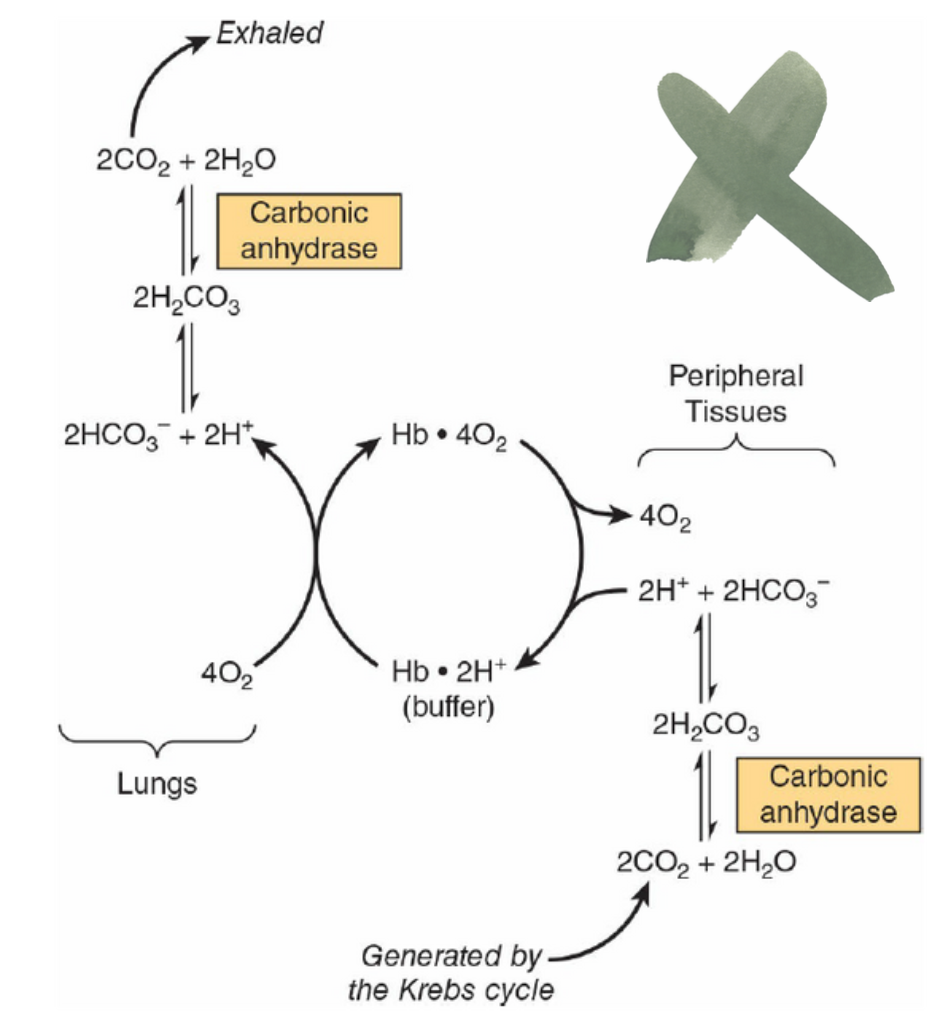
- ボーア効果: CO2とH2CO3の相互変換により、ヘモグロビンのT状態(oxygen-no-so-loving)とR状態(oxygen-so-loving)におけるプロトン(H+)と酸素(O2)の結合がそれぞれ逆転します。
- ハルダイン効果(Haldane Effect): ハルダイン効果は、HbのH+およびCO2に対する親和性が、Hb-O2飽和度の変化によってどのように影響を受けるかを説明します。
| 特徴 | ボーア効果(Bohr Effect) | ヘルダン効果(Haldane Effect) |
|---|---|---|
| 定義 | 酸素親和性の低下 血液中の二酸化炭素濃度が増加し、pHが低下することに応じて発生する。 | 二酸化炭素親和性の低下 血液中の酸素濃度が増加し、pHが上昇することに応じて発生する。 |
| 最初に記述した人物 | クリスチャン・ボーア(Christian Bohr) | ジョン・スコット・ヘルダン(John Scott Haldane) |
| 発生場所 | 代謝が活発な組織(Metabolizing tissue) | 肺(Lungs) |
| 効果 | 酸素の放出を促進 酸素が代謝組織で放出される。 | 二酸化炭素の放出を促進 二酸化炭素が肺で放出される。 |
| 有効なpH状態 | 低pHの状態で効果的(Effective under low blood pH) | 高pHの状態で効果的(Effective under high blood pH) |
| 原因 | 代謝組織での二酸化炭素の取り込みによって引き起こされる。 | 肺での酸素の取り込みによって引き起こされる。 |
| 役割 | 代謝組織での酸素放出を促進。 | ヘモグロビンへの酸素結合を促進。 |
その他の要因
- 2,3-ビスホスホグリセリン酸(BPG): CO2が赤血球のpHを低下させるときに生成されます。T状態のヘモグロビンはBPG分子を一つ結合し、塩橋を形成します。これにより、酸素化されていないヘモグロビンが安定化され、R状態への変換前に追加の塩橋が形成されます。
臨床的意義(Clinical Significance)
- ヘモグロビン異常症(Hemoglobinopathies)
- メトヘモグロビン(Methemoglobin)およびヘモグロビンM(Hemoglobin M): ヘム鉄がフェリック(Fe3+)状態となり、酸素を結合または輸送できなくなります。ヘモグロビンMでは、His F8がチロシンに置換され、Fe3+状態を安定化させる強力なイオン複合体を形成します。
- ヘモグロビンS(Hemoglobin S): HbSでは、非極性アミノ酸のバリンが、β鎖の表面にある極性残基Glu6に置き換わり、β鎖の表面に疎水性の「粘着パッチ」が生成されます。低酸素分圧では、脱酸素ヘモグロビンS(deoxyHbS)が長くて不溶性の繊維を形成することができます。脱酸素ヘモグロビンA(deoxyHbA)の結合により、HbAが別のヘモグロビン分子と結合するために必要な第二の粘着パッチを欠いているため、繊維の重合が停止します。これらのねじれた螺旋状の繊維は赤血球を特徴的な鎌状に変形させ、脾臓の洞隙において溶解しやすくします。
- ミオグロビン尿症(Myoglobinuria): 骨格筋に大量の外傷が加わり、それが腎臓障害を引き起こします。
- 貧血(Anemia): 赤血球やヘモグロビンの減少。鉄欠乏症、赤血球産生の低下、ビタミンB12欠乏症などが原因です。
- サラセミア(Thalassemias): ヘモグロビンのαまたはβ鎖の一部または全体の欠失による遺伝的欠陥です。
- 一酸化炭素中毒(Carbon Monoxide Poisoning): 一酸化炭素(CO)は無臭無色のガスで、化石燃料の燃焼時に発生します。COは肺でほぼ変化なく吸収され、吸収後、主にヘモグロビンに結合します(90%)。一部はミオグロビンやシトクロムCオキシダーゼに結合し、1%未満が血漿に溶解し、また1%未満が二酸化炭素に酸化されます。
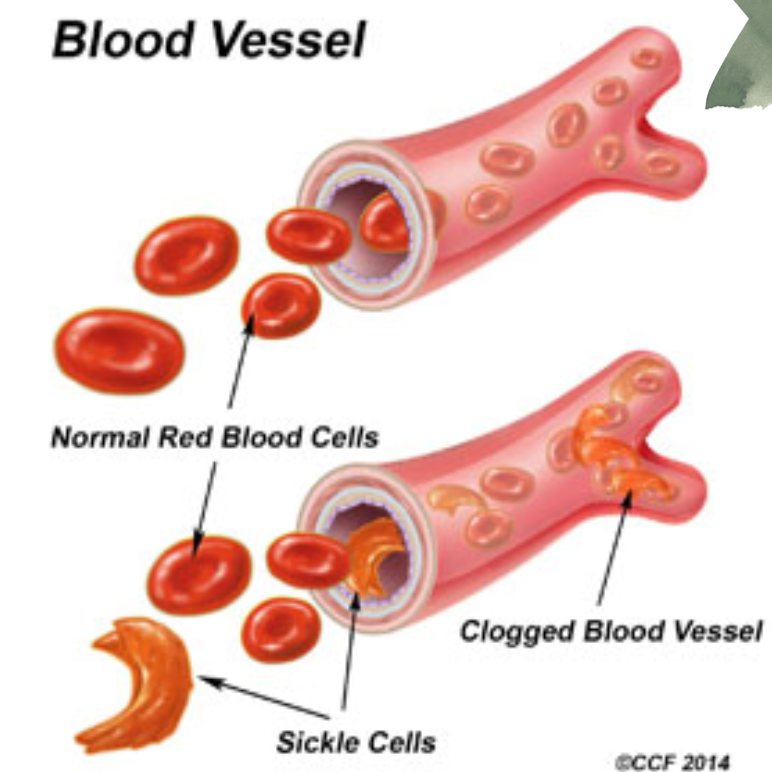
ミオグロビン尿症(Myoglobinuria)
- ミオグロビン尿症は、大量の骨格筋損傷や外傷が原因で、筋肉から放出されたミオグロビン(myoglobin)が腎臓に到達し、尿中に排出される状態です。筋肉損傷後に発生し、尿が赤褐色に変色することがあります。腎臓がミオグロビンを処理できない場合、腎障害を引き起こす可能性があります。
貧血(Anemia)
- 鉄欠乏性貧血(Iron Deficiency Anemia)
- 概要: 鉄(Fe)が不足することで、ヘモグロビンの合成が不十分になり、赤血球が正常な大きさと形状で作られなくなります。
- 原因: 鉄分不足、吸収不良、慢性的な出血(例: 消化管出血)。
- 症状: 疲労感、息切れ、めまい、皮膚の蒼白など。
- ビタミンB12欠乏性貧血(Vitamin B12 Deficiency Anemia)
- 概要: ビタミンB12が不足すると、赤血球が正常に成熟せず、大きく異常な形状の赤血球が生成されます(巨赤芽球性貧血)。
- 原因: 食事からの不足、吸収不良(例: 胃切除、悪性貧血)。
- 症状: 疲労感、息切れ、手足のしびれやむくみ、記憶障害など。
- 鉛中毒(Lead Poisoning)
- 概要: 鉛が体内に蓄積し、赤血球の合成や機能に悪影響を与えます。鉛は鉄の代わりにヘムの合成に関与し、正常な赤血球が作られなくなります。
- 原因: 鉛を含む塗料、配管、工業製品などの長期間の接触。
- 症状: 貧血、腹痛、頭痛、神経系の障害(例: 記憶障害、運動失調)など。
サラセミア(Thalassemias)と他のヘモグロビン異常症(Hemoglobinopathies)
- αサラセミア(Alpha Thalassemia)とβサラセミア(Beta Thalassemia)
- 概要: ヘモグロビンのα鎖またはβ鎖のいずれかが欠損または異常である遺伝的疾患です。
- αサラセミア: α鎖が欠損または変異し、異常なヘモグロビンが生成されます。軽度から重度までの症状があり、重症型では胎児期から症状が現れることがあります。
- βサラセミア: β鎖が欠損または変異し、異常なヘモグロビンが生成されます。症状は軽度から重度まであり、重症型では定期的な輸血が必要です。
- 症状: 貧血、黄疸、骨の変形、成長障害など。
- ヘモグロビン異常症(Hemoglobinopathies)
- 概要: ヘモグロビンのアミノ酸配列に変異があり、正常なヘモグロビンが生成されない遺伝的疾患です。主要なものには、鎌状赤血球症(Sickle Cell Anemia)やヘモグロビンC病(Hemoglobin C Disease)があります。
- 鎌状赤血球症(Sickle Cell Anemia)
- 概要: β鎖の第6番目のアミノ酸がグルタミン酸からバリンに置き換わることで、赤血球が異常な鎌状に変形します。
- 症状: 鎌状の赤血球が血管を閉塞させることで、疼痛発作、貧血、感染症リスクの増加、臓器障害などが発生します。
- ヘモグロビンC病(Hemoglobin C Disease)
- 概要: β鎖の第6番目のアミノ酸がグルタミン酸からリジンに置き換わることで発生します。症状は比較的軽度で、ヘモグロビンCの存在が血液検査で確認されます。
- 症状: 軽度の貧血、脾臓の肥大など。
一酸化炭素中毒(Carbon Monoxide Poisoning)
- 概要: 一酸化炭素(CO)は無臭無色のガスで、燃焼過程で生成されます。吸入するとヘモグロビンに結合し、酸素の結合部位を占有することで、酸素の運搬能力が低下します。
- 症状: 頭痛、めまい、吐き気、意識障害、重篤な場合には死亡することもあります。
高山病(Altitude Sickness)
- 概要: 高地に急激に移動した際に発生する病気で、酸素濃度が低い環境で身体が適応できなくなります。
- 症状: 頭痛、吐き気、めまい、息切れ、重症の場合は肺水腫や高山脳症が発生することがあります。適切な休息と酸素供給が必要です。
Proteins: Myoglobin & Hemoglobin
OBJECTIVES
After studying this chapter, you should be able to:
■ Describe the most important structural similarities and differences between myoglobin and hemoglobin.
■ Sketch binding curves for the oxygenation of myoglobin and hemoglobin.
■ Explain why hemoglobin’s sigmoidal O2-binding curve renders it a better
vehicle for transporting oxygen than myoglobin’s hyperbolic binding curve.
■ Explain how the presence of the distal histidine affects the ability of hemoglobin to bind carbon monoxide (CO).
■ Define P50 and indicate its significance in oxygen transport and delivery.
■ Describe the structural and conformational changes in hemoglobin that
accompany its oxygenation and subsequent deoxygenation. What role does the proximal histidine play in this transition?
■ Explain the role of 2,3-bisphosphoglycerate (BPG) in oxygen binding and delivery.
■ Explain how the Bohr effect a) enhances the ability of red blood cells to absorb CO2 from peripheral tissues and b) promotes the release of transported CO2 in the O2 rich environment of the lungs.
■ Describe the structural consequences to hemoglobin S (HbS) of lowering Po2.
■ Identify the metabolic defect that occurs as a consequence of α and β thalassemias.
BIOMEDICAL IMPORTANCE
The efficient delivery of oxygen from the lungs to the peripheral tissues and the maintenance of tissue reserves to protect against anoxic episodes are essential to health. In mammals, these functions are performed by the homologous heme proteins hemoglobin and myoglobin, respectively. Myoglobin, a monomeric protein of red muscle, binds oxygen tightly as a reserve against oxygen deprivation. The multiple subunits of hemoglobin, a tetrameric protein of erythrocytes, interact in a cooperative fashion that enables this transporter to offload a high proportion of bound O, in peripheral tissues while simultaneously retaining the capacity to bind it efficiently in the lungs. The offloading of oxygen is enhanced by the binding of 2,3-bisphosphoglycerate (BPG), which stabilizes the quaternary structure of deoxyhemoglobin. Cyanide and carbon monoxide (CO) kill because they disrupt the physiologic function of the heme proteins cytochrome oxidase and hemo-globin, respectively.
In addition to delivering O, hemoglobin plays a critical role in the transport of the waste product of respiration, COy to the lungs for disposal. Proton binding to hemoglobin enhances the conversion of CO,, a major product of respira-tion, into water-soluble carbonic acid and its conjugate base, bicarbonate by the enzyme carbonic anhydrase. In the lungs, O,-induced proton release from hemoglobin reverses this process to facilitate disposal as gaseous CO, Additional CO, is carried in the form of covalently bound carbamates. Hemoglobin and myoglobin illustrate both protein structure-function relationships and the molecular basis of genetic disorders such as sickle cell disease and the thalassemias.
HEME & FERROUS IRON CONFER THE ABILITY TO STORE & TRANSPORT OXYGEN
Myoglobin and hemoglobin contain heme, an iron-containing cyclic tetrapyrrole consisting of four molecules of pyrrole linked by methyne bridges. This planar network of conjugated double bonds absorbs visible light and colors heme deep red.
The substituents at the 3-positions of heme are methyl (M), vinyl (V), and propionate (Pr) groups arranged in the order
M, V, M, V, M, Pr, Pr, M (Figure 6-1). An atom of ferrous iron (Fe2+) resides at the center of the planar tetrapyrrole. Oxidation of the Fe2t of myoglobin or hemoglobin to Fest destroys their biologic activity. Other proteins with metal-containing tetra-pyrrole prosthetic groups include the cytochromes (Fe and Cu) and chlorophyll (Mg) (see Chapter 31).
Myoglobin Is Rich in a Helix
Oxygen stored in red muscle myoglobin is released during O, deprivation (eg, severe exercise) for use in muscle mitochondria for aerobic synthesis of ATP ( see Chapter 13).
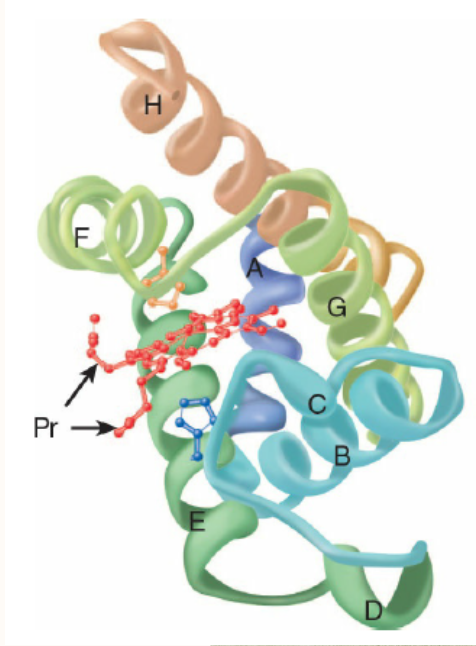
A 153-aminoacyl residue polypeptide (MW 17,000), the compactly folded myoglobin molecule measures 4.5 × 3.5 x 2.5 nm (Figure 6-2). An unusually high proportion, about
75%, of the residues are present in eight right-handed 7 to
20 residue a helices. Starting at the amino terminal, these are termed helices A through H. Typical of globular pro-teins, the surface of myoglobin is rich in amino acids bearing polar and potentially charged side chains, while-with two exceptions-the interior contains residues that possess nonpolar R groups (eg, Leu, Val, Phe, and Met). The exceptions are the seventh and eighth residues in helices E and F, His E7 and His F8, which lie close to the heme iron, the site of O, binding.
Histidines F8 & E7 Perform Unique Roles in Oxygen Binding
The heme of myoglobin lies in a crevice between helices E and F oriented with its polar propionate groups facing the surface of the globin (see Figure 6-2). The remainder resides in the nonpolar interior. It is held in place mainly by hydrophobic interactions. The fifth coordination position of the iron is occupied by a nitrogen from the imidazole ring of the proximal his-tidine, His F8. The distal histidine, His E7, lies on the side of the heme ring opposite to His F8.
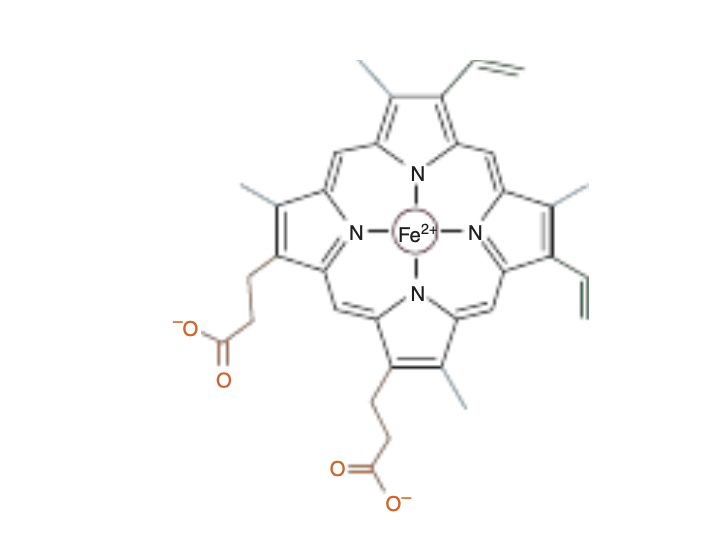
FIGURE6–1 Heme.Thepyrroleringsandmethynebridgecar- bons are coplanar, and the iron atom (Fe2+) resides in almost the same plane. The fifth and sixth coordination positions of Fe2+ are directly perpendicular to—and directly above and below—the plane of the heme ring. Observe the nature of the methyl (blue), vinyl (green), and propionate (orange) substituent groups on the β carbons of the pyr- role rings, the central iron atom (red), and the location of the polar side of the heme ring (at about 7 o’clock) that faces the surface of the myoglobin molecule.
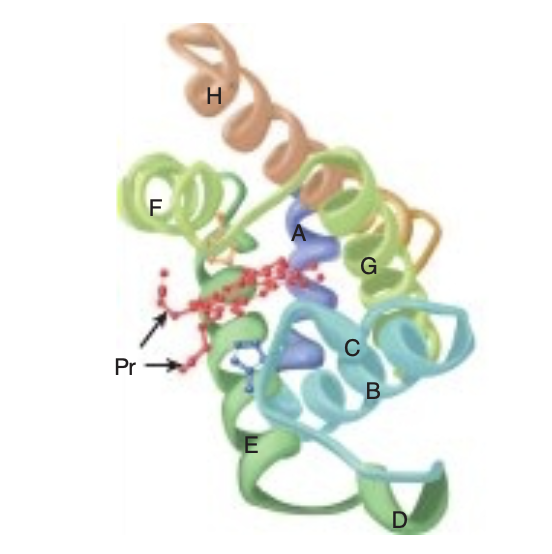
FIGURE 6-2 Three-dimensional structure of myoglobin.
Shown is a ribbon diagram tracing the polypeptide backbone of myo-globin. The color of the polypeptide chain is graded along the visible spectrum from blue (N-terminal) to tan (C-terminal). The heme prosthetic group is red. The a-helical regions are designated A through H. The distal (E7) and proximal (F8) histidine residues are highlighted in blue and orange, respectively. Note how the polar propionate sub-stituents (Pr) project out of the heme toward solvent. (Adapted with permission from Protein Data Bank ID no. 1a6n.)
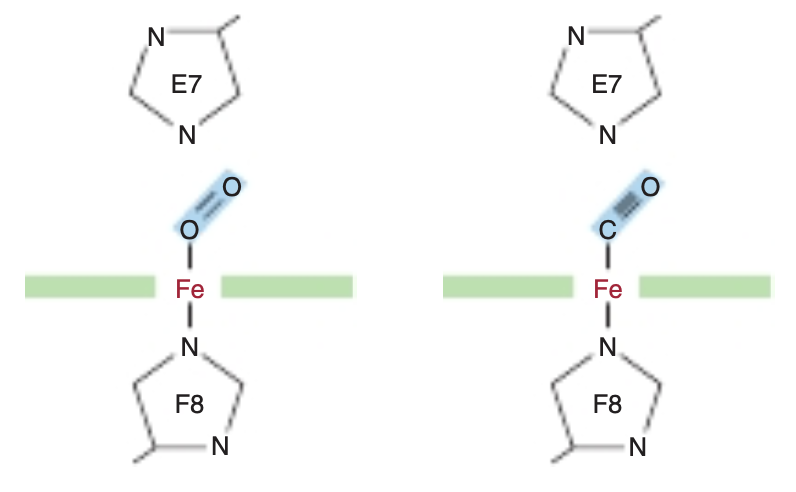
FIGURE 6-3 Angles for bonding of oxygen and carbon monoxide (CO) to the heme iron of myoglobin. The distal E7 histidine hinders bonding of CO at the preferred (90°) angle to the plane of the heme ring. The Iron Moves Toward the Plane of the Heme When Oxygen Is Bound The iron of unoxygenated myoglobin lies 0.03 nm (0.3 A) outside the plane of the heme ring, toward the proximal histidine, His F8. Consequently, the heme “puckers” slightly. When O, occupies the sixth coordination position, the iron moves to within 0.01 nm (0.1 À) of the plane of the heme ring.
Oxygenation of myoglobin thus is accompanied by motion of the iron, of His F8, and of residues linked to His F8.
Apomyoglobin Provides a Hindered Environment for the Heme Iron
Minute quantities of CO arise from a variety of biologic sources, including the catabolism of red blood cells within the human body. CO is also present in the atmosphere, mainly due to the incomplete combustion of fossil fuels. CO binds to the iron in a free heme group 25,000 times more strongly than oxygen. However, the heme groups in the apoproteins of myoglobin and hemoglobin reside in a hindered environment for their gaseous ligands (Figure 6-3). When CO binds to free heme, all three atoms (Fe, C, and O) lie perpendicular to the plane of the heme. This geometry maximizes the overlap between the lone pair of electrons on the sp hybridized carbon of the CO molecule and the Fett iron (Figure 6-4, right). By contrast, O. is sp? hybridized. Consequentlv, the electrons that bind to the iron lie at an angle of roughly 120° to the axis of the 0=0 double bond (see Figure 6-4, left). In myoglobin and hemoglobin the distal histidine sterically precludes or hinders the preferred, high-affinity orientation of CO while still permitting O, to attain its most favorable orientation (see Figure 6-3). Binding at a less favored angle reduces the strength of the heme-CO bond to about 200 times that of the heme-O, bond (see Figure 6-3, right). Therefore O,, which typically is present in great excess over CO, normally dominates.
Nevertheless, about 1% of human myoglobin typically is present combined with CO.
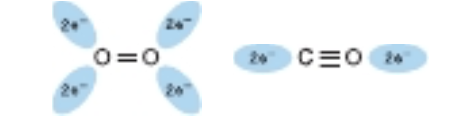
FIGURE 6-4 Orientation of the lone pairs of electrons relative to the 0=0 and C=0 bonds of oxygen and carbon monoxide. In molecular oxygen, formation of the double bond between the two oxygen atoms is facilitated by the adoption of an sp? hybridization state by the valence electron of each oxygen atom. As a consequence, the two atoms of the oxygen molecule and each lone pair of electrons are coplanar and separated by an angle of roughly 120° (left). By contrast, the two atoms of carbon monoxide are joined by a triple bond, which requires that the carbon and oxygen atoms adopt an sp hybridization state. In this state, the lone pairs of electrons and triple bonds are arranged in a linear fashion, where they are separated by an angle of 180° (right).
THE OXYGEN DISSOCIATION CURVES FOR MYOGLOBIN & HEMOGLOBIN SUIT THEIR PHYSIOLOGIC ROLES
The hyperbolic relationship between the concentration, or partial pressure, of O, (Poy) and the quantity of O, bound, expressed as an O, saturation isotherm (Figure 6-5), reveals why myoglobin is more suitable for O, storage than for O, transport. Myoglobin loads O, readily at the PO, of the lung capillary bed (100 mm Hg). However, since myoglobin releases only a small fraction of its bound O, at the PO, values typically encountered in active muscle (20 mm Hg) or other tissues (40 mm Hg), it represents an ineffective vehicle for delivery of O, However, when strenuous exercise lowers the Po, of muscle tissue to about 5 mm of Hg, dissociation of 0, from myoglobin permits mitochondrial synthesis of ATP, and hence muscular activity, to continue.
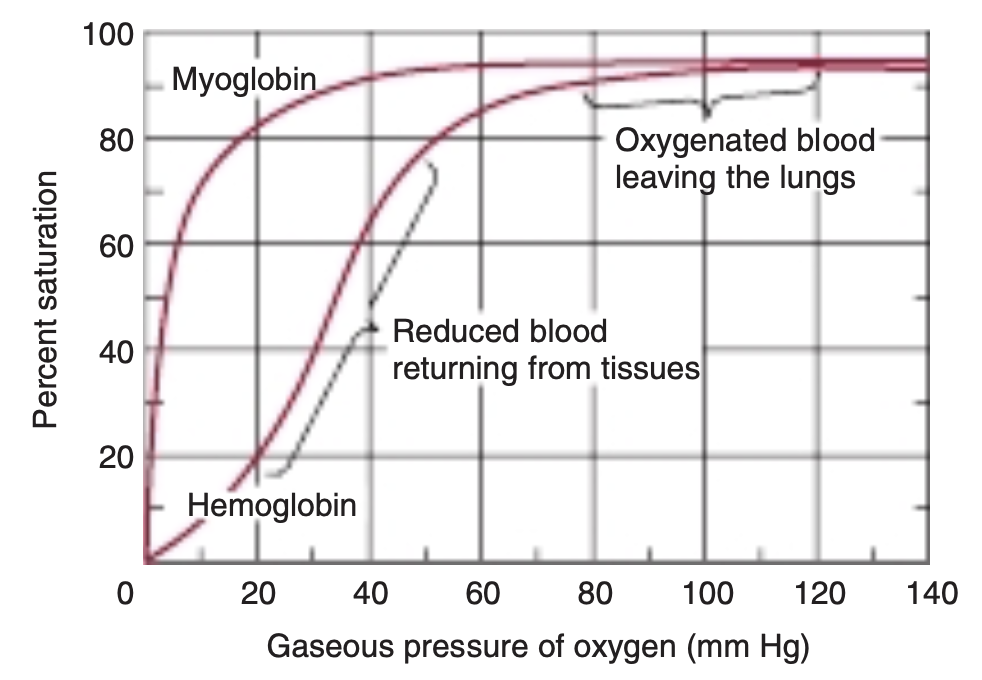
FIGURE 6-5 Oxygen-binding curves of both hemoglobin and myoglobin. Arterial oxygen tension is about 100 mm Hg; mixed venous oxygen tension is about 40 mm Hg; capillary (active muscle) oxygen tension is about 20 mm Hg; and the minimum oxygen tension required for cytochrome oxidase is about 5 mm Hg. Association of chains into a tet-rameric structure (hemoglobin) results in much greater oxygen delivery than would be possible with single chains. (Modified with permission from Scriver CR, Beaudet AL, Sly WS: The Molecular and Metabolic Bases of Inherited Disease, 7th ed. New York, NY: McGraw Hill; 1995.)
By contrast, hemoglobin behaves as if it were two proteins.
At the high Po, of the lungs, 100 mm Hg and above, it displays a high affinity for oxygen that enables it to bind oxygen to nearly every available heme iron. This form of the protein is commonly referred to as R, for relaxed, state hemoglobin. At the lower PO, values encountered in peripheral tissues, 40 mm Hg and below, hemoglobin exhibits a much lower apparent affinity for oxygen.
Transition of hemoglobin to this low affinity, taut or T state enables it to release a large proportion of the oxygen previously picked up in the lungs. This dynamic interchange between the high and low affinity R and T states serves as the foundation for hemoglobins sigmoidal O,-binding curve.
THE ALLOSTERIC PROPERTIES OF HEMOGLOBINS RESULT FROM THEIR QUATERNARY STRUCTURES
The quaternary structure of hemoglobin confers striking additional properties, absent from monomeric myoglobin, which adapts this tetrameric protein to its unique biologic roles in the reciprocal transport of O, and CO, between the lungs and peripheral tissues.
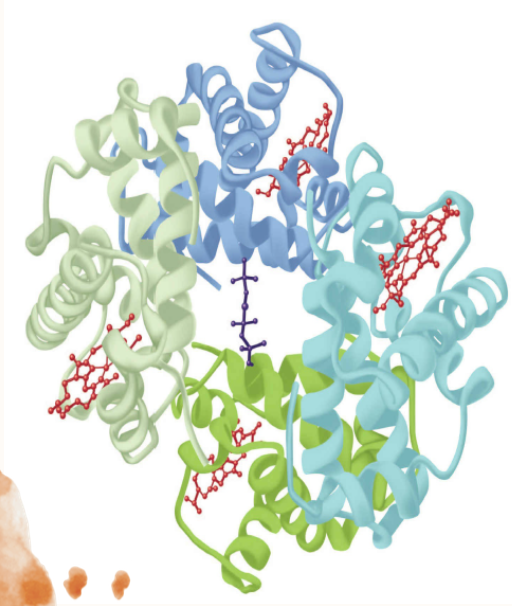
Hemoglobin Is Tetrameric
Hemoglobins are tetramers composed of pairs of two similar, but distinct, polypeptide subunits (Figure 6-6). Greek letters are used to designate each subunit type. The subunit composition of the principal hemoglobins are a,, (HbA; normal adult hemoglobin), aY, (HbF; fetal hemoglobin), a,B, (HbS; sickle cell hemoglobin), and a,S, (HbA,; a minor adult hemoglobin).
The primary structures of the B, y, and 8 chains of human hemoglobin are highly conserved.
Myoglobin & the ß Subunits of Hemoglobin Share Almost Identical Secondary & Tertiary Structures
Despite differences in the kind and number of amino acids present, myoglobin and the B polypeptide of hemoglobin A share almost identical secondary and tertiary structures.
Similarities include the location of the heme and the helical
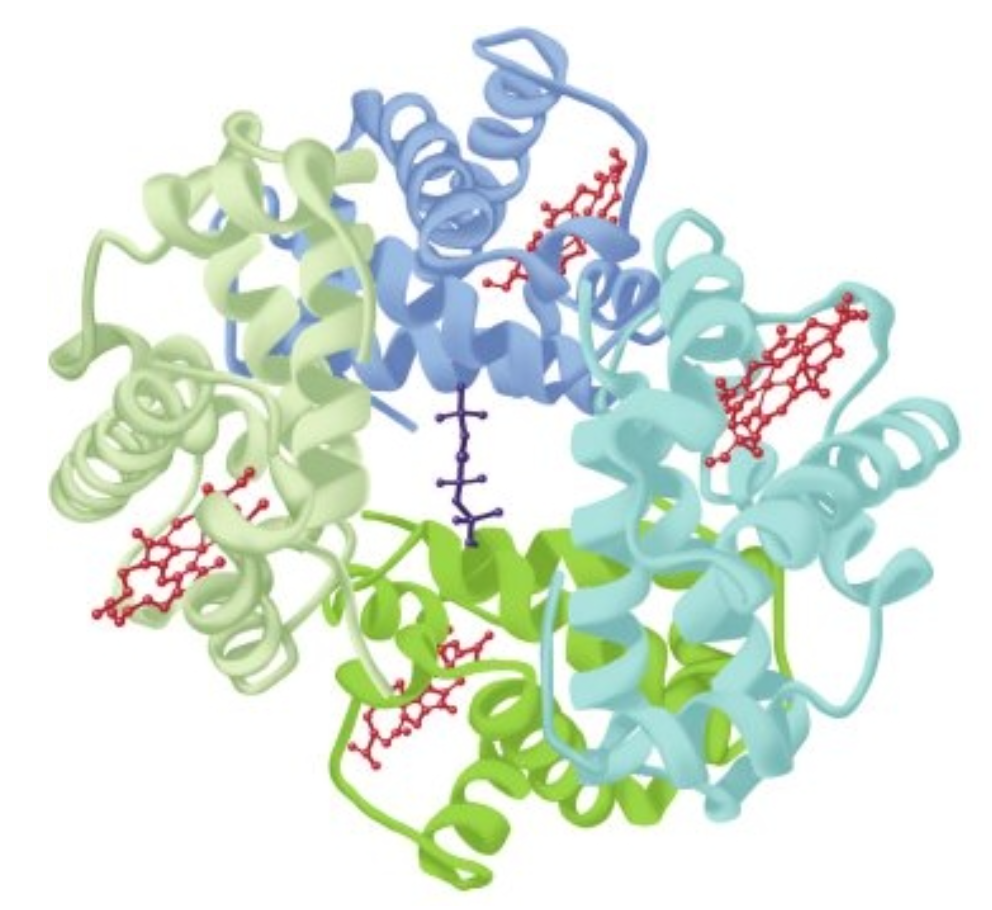
FIGURE6–6 Hemoglobin.Shownisthethree-dimensionalstructureofdeoxyhemoglobinwithamoleculeof2,3-bisphosphoglycerate (dark blue) bound. The two α subunits are colored in the darker shades of green and blue, the two β subunits in the lighter shades of green and blue, and the heme prosthetic groups in red. (Adapted with permission from Protein Data Bank ID no. 1b86.)
regions, and the presence of amino acids with similar properties at comparable locations. Although it possesses seven rather than eight helical regions, the a polypeptide of hemoglobin also closely resembles myoglobin.
Oxygenation of Hemoglobin Triggers Conformational Changes in the Apoprotein
Hemoglobins can bind up to four molecules of O, per tetramer, one per heme. However, once the first molecule of O, becomes bound, the affinity of the other three subunits increases (see Figure 6-5). Termed cooperative binding, this behavior permits hemoglobin to maximize both the quantity of O, loaded at the Po, of the lungs and the quantity of O, released at the Po, of the peripheral tissues.
P. Expresses the Relative Affinities of Different Hemoglobins for Oxygen
The quantity P so a measure of O, concentration, is the partial pressure of O, at which a given hemoglobin reaches half-saturation. Depending on the organism, Ps can vary widely, but in all instances, it exceeds the normal Po, of the peripheral tissues. For example, the values of P. for HbA and HbF are 26 and 20 mm Hg, respectively. In the placenta, this difference enables HbF to extract oxygen from the HbA in the mother’s blood. However, HbF is suboptimal postpartum since its higher affinity for O, limits the quantity of O, delivered to the tissues.
The subunit composition of hemoglobin tetramers undergoes complex changes during development. The human fetus initially synthesizes a E&, tetramer. By the end of the first tri-mester, § and a subunits have been replaced by a and y sub-units, forming HbF (a,y,), the hemoglobin of late fetal life.
While synthesis of B subunits begins in the third trimester, the replacement of y subunits by B subunits to yield adult HbA (a,B,) does not reach completion until several weeks postpartum (Figure 6-7).
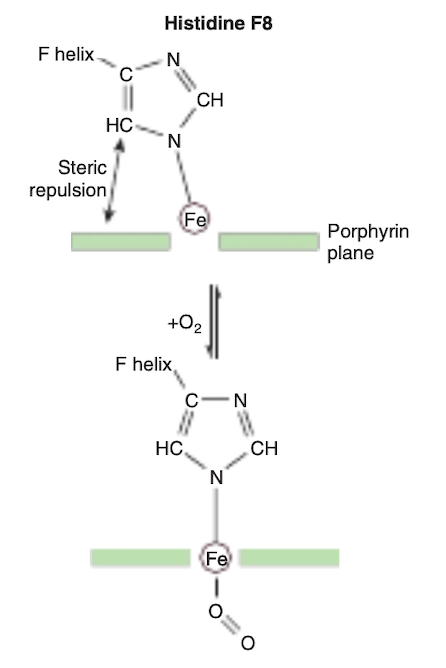
FIGURE 6-8 On oxygenation of hemoglobin the iron atom moves into the plane of the heme. Histidine F8 and its associated aminoacyl residues are pulled along with the iron atom. For a representation of this motion, see https://pdb101.rcsb.org/learn/videos/ oxygen-binding-in-hemoglobin.
Oxygenation of Hemoglobin Is Accompanied by Large Conformational Changes
The binding of the first molecule of O, to deoxyHb shifts the heme iron toward the plane of the heme ring (Figure 6-8).
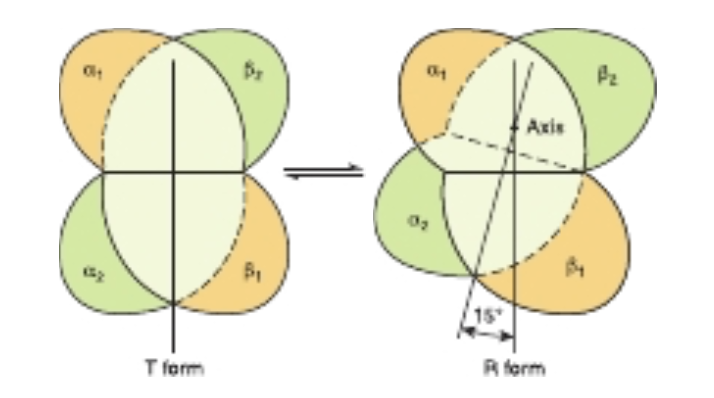
FIGURE 6-9 During transition of the T form to the R form of hemoglobin, the a P, pair of subunits (green) rotates through 15° relative to the pair of a,, subunits (yellow). The axis of rotation is eccentric, and the a,B, pair also shifts toward the axis somewhat. In the representation, the tan a,B, pair is shown fixed while the green a,B, pair of subunits both shifts and rotates.
This motion is transmitted through the proximal (F8) histidine and the residues attached thereto to the entire tetramer, triggering the rupture of salt bridges formed by the carboxyl terminal residues of all four subunits. As a result, one pair of a/B subunits rotates 15° with respect to the other, compacting the tetramer (Figure 6-9) and triggering other, profound changes in secondary, tertiary, and quaternary structures that accompany the transition of hemoglobin from the low-affinity T state to the high-affinity R state. These changes significantly increase the affinity of the remaining unoxygenated hemes for O,, as subsequent binding events require the rupture of fewer salt bridges (Figure 6-10). The cooperativity of hemoglobin constitutes a form of allosteric (Gk allos “other,” steros “space”) behavior (see Chapter 19), since binding of O, to one heme affects the binding affinity of the others.
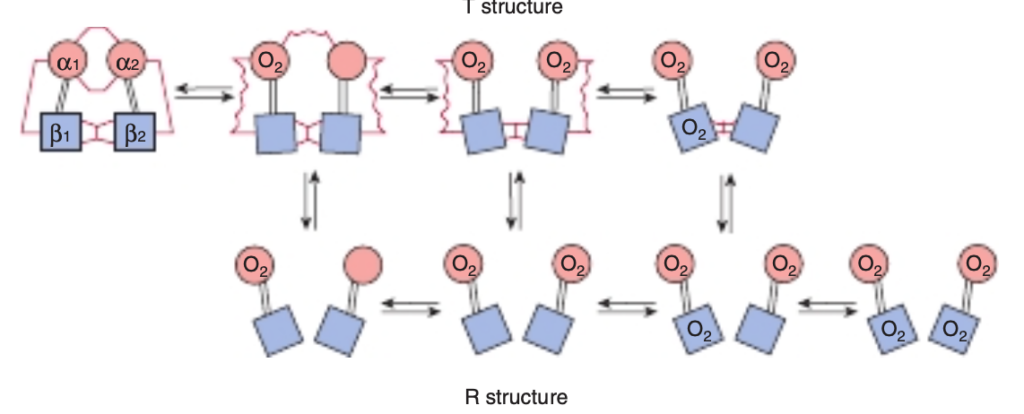
FIGURE 6-10 Transition from the T structure to the R structure. In this model, salt bridges (red lines) linking the subunits in the T structure break progressively as oxygen is added, and even those salt bridges that have not yet ruptured are progressively weakened (wavy red lines). The transition from T to R does not take place after a fixed number of oxygen molecules have been bound but becomes more probable as each successive oxygen binds. The transition between the two structures is influenced by protons, carbon dioxide, chloride, and 2,3 bisphosphoglycerate (BPG); the higher their concentration, the more oxygen must be bound to trigger the transition. Fully oxygenated molecules in the T structure and fully deoxygenated molecules in the R structure are not shown because they are unstable. (Modified with permission from Perutz MF. Hemoglobin structure and respiratory transport. Sci Am. 1978;239(6):92-125.)
Hemoglobin Assists in the Transport of CO, to the Lungs
For every molecule of O, consumed during the course of res-piration, one molecule of CO, is produced. Consequently, red blood cells must be able to efficiently absorb CO, from respiting tissues and subsequently release it for disposal in the lungs.
Two key contributors to this process are the enzyme carbonic anhydrase, which catalyzes the hydration of CO, to water-soluble carbonic acid, H,CO, and the ability of hemoglobin to cooperatively transition between the T and R states.
Carbonic Anhydrase Converts CO, to Water-Soluble Carbonic Acid & Bicarbonate
As is the case for O,, the limited water solubility of CO, at neutral pH renders simolv dissolving this gas in the bloodstream grossly insufficient to meet the body’s transport needs. The enzyme carbonic anhydrase, present in large quantities in red blood cells, catalyzes the hydration of CO, molecules to form water soluble carbonic acid, H,CO.
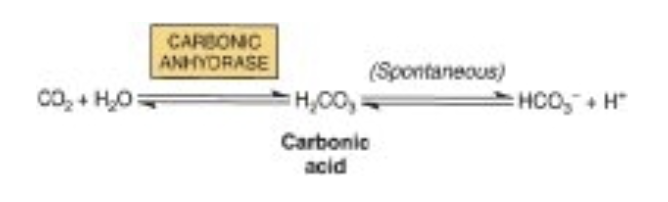
H,CO, is a weak acid that, in turn, can dissociate to form bicarbonate ion, HCO;,and a proton. Since the pK, of H, CO, 6.35, falls below physiologic pH, in red blood cells the majority of absorbed CO, is carried as water-soluble bicarbonate.
Binding of Protons to T-State Hemoglobin Increases CO, Uptake From Respiring Tissues
When hemoglobin transitions from the R to the T state, conformational changes take place in each of the two B-chains that lead to the formation of salt bridges between the side chains of Asp 94 and His 146. Formation of each salt bridge requires T-state hemoglobin to bind a proton from the surrounding environment. As R-state hemoglobin gives up its bound O, to respiring tissues and subsequently transitions to the T state, the absorption of these two protons both buffers the pH of the acidifying red blood cell and shifts or pulls the equilibrium between H,CO, and HCO, in favor of bicarbon-ate, further increasing the quantity of carbon dioxide absorbed by the red blood cells from peripheral tissues.
T-state hemoglobin binds two protons per tetramer. In actively respiring tissues, the proton concentration inside the red blood cells will increase as H,CO, accumulates. The greater availability of H*, in turn, favors the formation of T-state hemoglobin, thereby enhancing the release of 0,.
Proton binding by T-state hemoglobin enables the high levels of CO, in actively respiring tissues to drive the release of 0, from hemoglobin while concomitantly enhancing the quantity of CO, absorbed by promoting the conversion of carbonic acid to bicarbonate. As a result, transition to the T state helps buffer or moderate the CO -mediated decrease in the pH of the red blood cells in venous blood.
Proton Release From R-state Hemoglobin Enhances CO, Release in the Lungs
On reaching the lungs, the dramatic increase in the partial pressure of oxygen drives the binding of O, to deoxyhemo-globin. O, binding, in turn, triggers the transition of hemoglobin from the T to the R state. The resulting conformational change causes the rupture of salt bridges in the two B-chains and the release of the two protons formerly chelated between Asp 94 and His 146. Once freed, the protons combine with bicarbonate to increase the concentration of carbonic acid, which in turns favors the carbonic anhydrase catalyzed dehydration of H,CO, to form CO,, which can then be disposed by exhalation (Figure 6-11). This proton-mediated coupling of the transition of hemoglobin between T and R states with the equilibria between CO, H,CO, and HCO, is known as the Bohr effect.
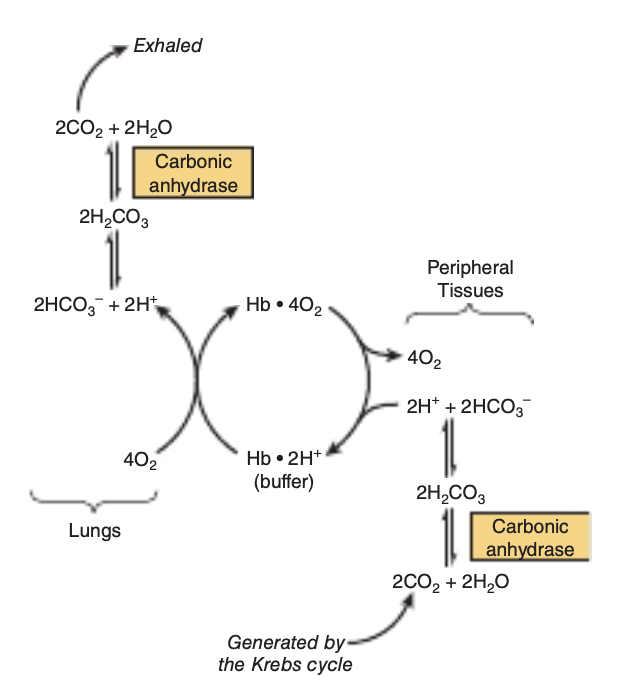
FIGURE 6-11 The Bohr effect. Carbon dioxide generated in peripheral tissues combines with water to form carbonic acid, which dissociates into protons and bicarbonate ions. Deoxyhemoglobin acts as a buffer by binding protons and delivering them to the lungs. In the lungs, the uptake of oxygen by hemoglobin releases protons that combine with bicarbonate ion, forming carbonic acid, which when dehydrated by carbonic anhydrase becomes carbon dioxide, which then is exhaled.
Additional CO, Is Transported as Carbamates of Hemoglobin
About 15% of the CO, in venous blood is carried by hemoglobin as carbamates formed with the amino terminal nitrogens of its polypeptide chains:
H I
COz + Hb-NHz* = 2H* + Hb-
N° C-0-
Carbamate formation changes the charge on amino terminals from positive to negative, favoring salt bridge formation between a and B chains. As part of the Bohr effect, proton binding by T-state hemoglobin favors carbamate formation while proton release on transition to the R state favors carbamate breakdown and release of CO,.
2,3-BPG Stabilizes the T Structure of Hemoglobin
The transition of hemoglobin to the T state opens a central cavity at the interface of its four subunits capable of binding one molecule of 2,3-bisphosphoglycerate, 2,3-BPG (see
Figure 6-6). Binding of 2,3-BPG thus favors the shift from the R to the I state and the further release of bound O, as conversion back to the R state requires the rupture of the additional salt bridges formed between 2,3-BPG and Lys EF6, His H21, and the terminal amino groups of Val NA1 from both B chains (Figure 6-12).
2,3-BPG is synthesized from the glycolytic intermediate 1,3-BPG, a reaction catalyzed by the bifunctional enzyme 2,3-bisphosphogylcerate synthase/2-phosphatase (BPGM).
BPG is hydrolyzed to 3-phosphoglycerate by the 2-phosphatase
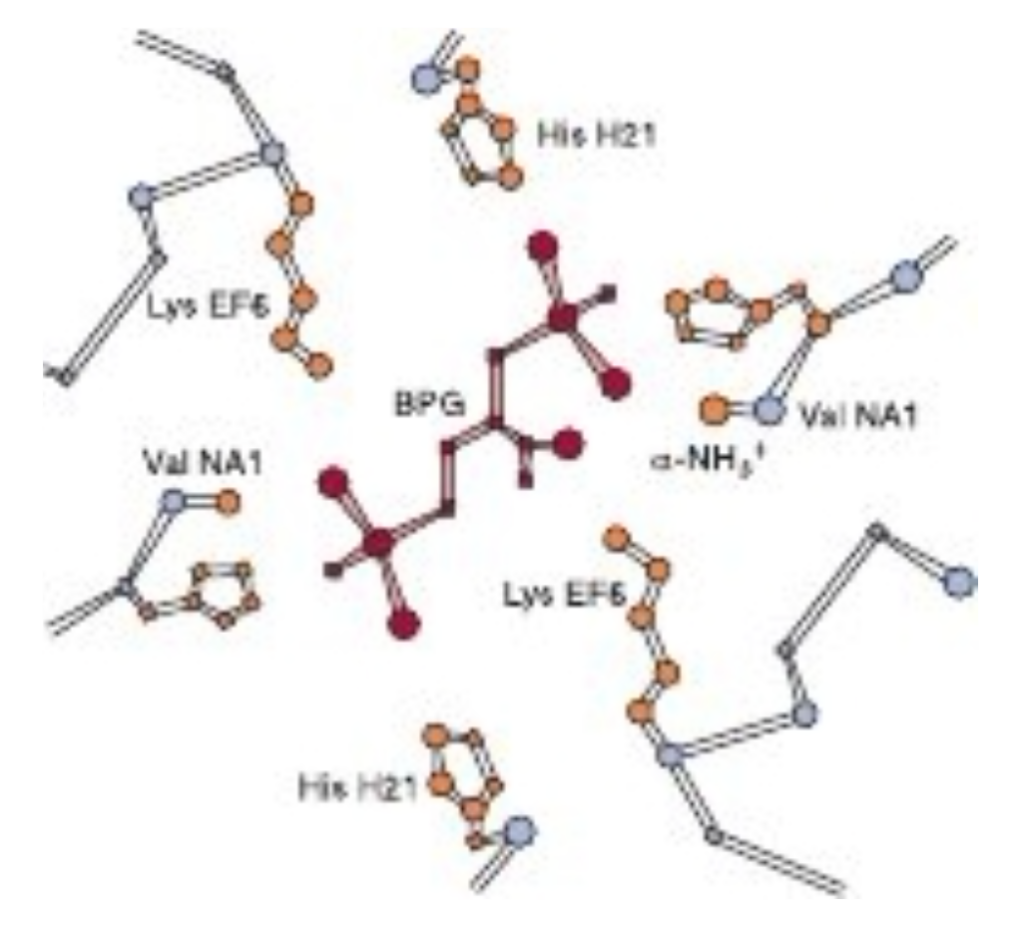
FIGURE6-12: Mode of binding of 2,3-bisphosphoglycerate (BPG) to human deoxyhemoglobin. BPG interacts with three positively charged groups on each β chain. (Adapted with permission from Arnone A. X-ray diffraction study of binding of 2,3-diphosphoglycerate to human deoxyhaemoglobin. Nature. 1972;237(5351):146-149.)
activity of BPGM and to 2-phosphoglycerate by a second enzyme, multiple inositol polyphosphate phosphatase (MIPP).
The activities of these enzymes, and hence the level of BPG in erythrocytes, are sensitive to pH. The CO, induced acidification of the red blood cells triggers production of 2,3-BPG, which in turn reinforces the impact of carbonic acid-derived protons in shifting the R-T equilibrium in favor of the I state, increasing the quantity of O, released in peripheral tissues.
In the fetal hemoglobin, residue H21 of the y subunit is Ser rather than His. Since Ser cannot form a salt bridge, BPG binds more weakly to HbF than to HbA. The lower stabilization afforded to the T state by BPG helps account for HbF having a higher affinity for O, than HbA.
Adaptation to High Altitude
Physiologic changes that accompany prolonged exposure to high altitude include increases in the number of ervthro-cytes, the concentration of hemoglobin within them, and the synthesis of 2,3-BPG. Elevated 2,3-BPG lowers the affinity of HbA for O, (increases Ps), which enhances the release of O, at peripheral tissues.
NUMEROUS MUTATIONS AFFECTING HUMAN HEMOGLOBINS HAVE BEEN DENTIFIED
Mutations in the genes that encode the a or subunits of hemoglobin potentially can affect its biologic function. However, of more than 1100 known genetic mutations affecting human hemoglobins, most are both rare and benign, presenting no clinical abnormalities. When a mutation does compromise biologic function, the condition is termed a hemoglobinopathy.
It is estimated that more than 7% of the globe’s population are carriers for hemoglobin disorders. The URL http://globin.cse. psu.edu/ (Globin Gene Server) provides information about-and links for-normal and mutant hemoglobins. Selected examples are as follows.
Methemoglobin & Hemoglobin M
In methemoglobinemia, the heme iron is ferric rather than ferrous, rendering methemoglobin unable to bind or transport Oz. Normally, the Fest of methemoglobin is returned to Fe?+ state through the action of the enzyme methemoglobin reductase. Methemoglobin levels can rise to pathophysiolog-ically significant levels from a number of causes: oxidation of Felt to Fe3t as a side effect of agents such as sulfonamides, reductions in the activity of methemoglobin reductase, or inheritance of the gene for a mutationally altered form of hemoglobin called H6M.
In hemoglobin M, histidine F8 (His F8) has been replaced by tyrosine. The iron of HbM forms a tight ionic complex with the phenolate anion of tyrosine that stabilizes the Fe3+ form. In a-chain hemoglobin M variants, the R-T equilibrium favors the T state. Oxygen affinity is reduced, and the Bohr effect is absent. B-Chain hemoglobin M variants exhibit R-T switch-ing, and the Bohr effect is therefore present.
Mutations that favor the R state (eg, hemoglobin Chesapeake) increase O, affinity. These hemoglobins therefore fail to deliver adequate O, to peripheral tissues. The resulting tissue hypoxia leads to polycythemia, an increased concentration of erythrocytes.
Hemoglobin S
In HbS, the nompolar amino acid valine has replaced the polar surface residue Glu6 of the subunit, generating a hydrophobic “sticky patch” on the surface of the B subunit of both oxyHbS and deoxyHbS. Both HbA and HbS contain a complementary sticky patch on their surfaces that is exposed only in the deoxygenated T state. Thus, at low Po,, deoxy HbS can polymerize to form long, twisted helical fibers. Binding of deoxy HbA terminates fiber polymerization, since HbA lacks the second sticky patch necessary to bind another Hb molecule (Figure 6-13). These insoluble fibers distort the erythrocyte into a characteristic sickle shape, rendering it vulnerable to lysis in the interstices of the splenic sinusoids. They also cause multiple secondary clinical effects. A low Po,, such as that at high altitudes, exacerbates the tendency to polymer-ize. The terms sickle cell trait and sickle cell disease refer to persons in whom the gene for either one or both & subunits are mutated, respectively. Emerging treatments for sickle cell disease include inducing HbF expression to inhibit the polymerization of HbS, stem cell transplantation, and, in the future, gene therapy.
BIOMEDICAL IMPLICATIONS
Myoglobinuria
Following massive crush injury to skeletal muscle followed by renal damage, released myoglobin may appear in the urine.
Myoglobin can be detected in plasma following a myocardial infarction, but assay of serum troponin, lactate dehydrogenase isozymes, or creatine kinase (see Chapter 7) provides a more sensitive index of myocardial injury.
Anemias
Anemias, reductions in the number of red blood cells or concentration of hemoglobin in the blood, can reflect impaired synthesis of hemoglobin (eg, in iron deficiency; see Chapter 52) or impaired production of erythrocytes (eg, in folic acid or vitamin B, deficiency; see Chapter 44). Diagnosis of anemias begins with spectroscopic measurement of blood hemoglobin levels.
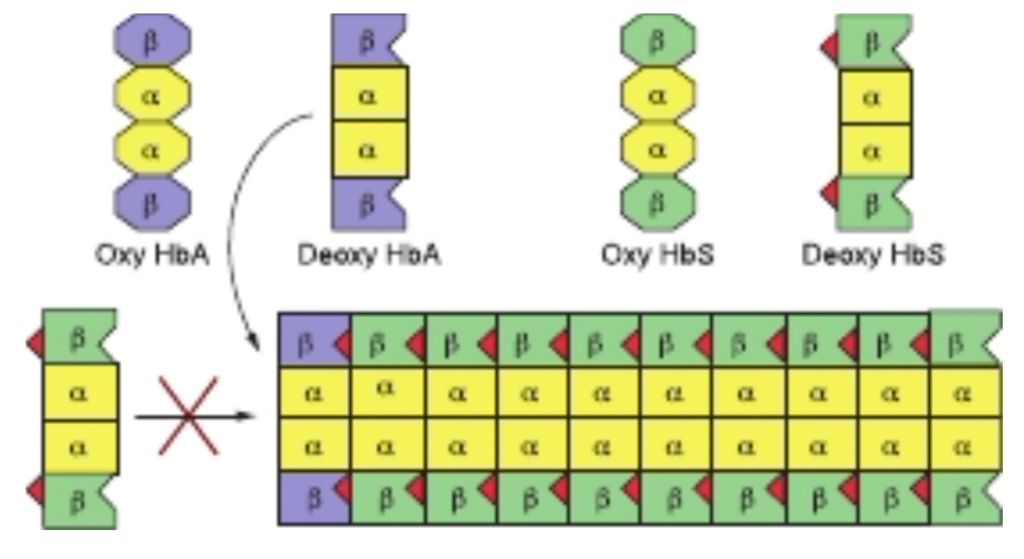
FIGURE 6-13 Polymerization of deoxyhemoglobin S. The dissociation of oxygen from hemoglobin S (HbS) unmasks a sticky patch (red triangle) on the surface of its § subunits (green) that can adhere to a complementary site on the subunits of other molecules of deoxyHbS. Polymerization to a fibrous polymer is interrupted deoxyHbA, whose subunits (lavender) lack the sticky patch required for binding additional HbS subunits.
Thalassemias
The genetic defects known as thalassemias result from the partial or total absence of one or more a or B chains of hemo-globin. Over 750 different mutations have been identified, but only three are common. Either the a chain (a thalassemias) or B chain (B thalassemias) can be affected. A superscript indicates whether a subunit is completely absent (a° or B° or whether its synthesis is reduced (a or -). Apart from marrow transplantation, treatment is symptomatic.
Certain mutant hemoglobins are common in many popu-lations, and a patient may inherit more than one type. Hemoglobin disorders thus present a complex pattern of clinical phenotypes. The use of DNA probes for their diagnosis is considered in Chapter 39.
GLYCATED HEMOGLOBIN (HBA,)
Blood glucose that enters the erythrocytes can form a covalent adduct with the &-amino groups of lysyl residues and the a-amino group of the N-terminal valines of hemoglobin B chains, a process referred to as glycation. Unlike glycosyl-ation (see Chapter 46), glycation is not enzyme-catalyzed. The fraction of hemoglobin glycated, normally about 5%, is proportionate to blood glucose concentration. Since the life-span of an erythrocyte is typically 120 days, the level of glycated hemoglobin (HbA) reflects the mean blood glucose concentration over the preceding 8 to 12 weeks. Measurement of HbA, therefore provides valuable information for management of diabetes mellitus.
SUMMARY
• Myoglobin is monomeric; hemoglobin is a tetramer of two subunit types (a,B, in HbA). Myoglobin and the subunits of hemoglobin are homologs of one another that, despite differences in primary structure, exhibit nearly identical secondary and tertiary structures.
- Heme is an essentially planar, slightly puckered, cyclic tetrapyrrole whose four nitrogen atoms bind a central Fe2+ that is also linked to histidine F8, and, in oxyMb and oxyHib, to Oz.
- The O,-binding curve for myoglobin is hyperbolic, but for hemoglobin it is sigmoidal, a consequence of cooperative interactions in the tetramer.
- Cooperativity arises from the ability of hemoglobin to exist in two different conformational states, a relaxed or R state in which all four subunits exhibit a high affinity for oxygen and a taut or T state where all four subunits display a low affinity for oxygen.
- The high levels of O, in the lungs drive the R-T equilibrium in favor of the R state, while acidification of the red blood cells generated from the catalytic hydration of CO, in the peripheral tissues favors the T state. Cooperativity thus maximizes the ability of hemoglobin both to load O, at the Po, of the lungs and to deliver O, at the Po, of the tissues.
- Relative affinities of different hemoglobins for oxygen are expressed as Pso the Po, at which they become half-saturated with O,. Hemoglobin isoforms saturate at the partial pressures of their respective respiratory organ, for example, the lung or placenta.
- On oxygenation of hemoglobin, the iron and histidine F8 move toward the heme ring. The resulting conformational changes in the hemoglobin tetramer induce the rupture of several salt bonds, the release of bound protons, and loosening of the quaternary structure that increases the affinity for the other three subunits for Oz
- Binding of 2,3-BPG in the central cavity of T-state hemoglobin favors O, release in actively respiring tissues. Transition to the R state results in the closure of this cavity and extrusion of 2,3-BPG.
- Hemoglobin assists in CO, transport from peripheral tissues to the lungs via the formation of carbamates and the Bohr effect, a consequence of the binding of protons to T-state, but not R-state, hemoglobin. Proton binding enhances the conversion of CO, to water-soluble carbonic acid and bicarbonate. In the lungs, the release of protons from oxygenated R-state hemoglobin favors the conversion of bicarbonate and carbonic acid to CO,, which is exhaled.
- In sickle cell hemoglobin (HbS), Val replaces Glu of the B subunit of HbA, creating a “sticky patch” that has a complement on deoxyHb (but not on oxy Hb). DeoxyHbS polymerizes at low O, concentrations, forming fibers that distort erythrocytes into sickle shapes.
- a and 3 Thalassemias are anemias that result from reduced production of a and B subunits of HbA, respectively.

コメント